3
Response of Articular Cartilage to Injury
VAHÉ R. PANOSSIAN
Articular cartilage is a homogeneous connective tissue that caps the articulating ends of synovial joints. Composed of primarily type II collagen, it is avascular and without innervation. It consists of a relatively small number of cells (chondrocytes) embedded in an abundance of extracellular matrix, composed of collagen, protein polysaccharides (proteoglycans), and water1,2 (Fig. 3–1). The physical properties of articular cartilage are determined by the chemical nature and arrangements of these constituents.3,4
Chondrocytes are responsible for the synthesis of the extracellular matrix. In the skeletally immature individual, chondrocytes control the growth and remodeling of the articular cartilage. In the adult, however, chondrocytes are oriented into four transitional zones, which include the tangential zone, transitional zone, radial zone, and the calcified zone (Fig. 3–2). The cells within these zones differ in spatial orientation, morphology, and force transduction.5–7
Since the beginnings of early orthopaedics, the perplexing nature of articular cartilage defects have been well recognized as difficult to treat.8–10 In 1743, Hunter11 stated that “from Hippocrates down to the present age, we shall find that ulcerated cartilage is universally allowed to be a very troublesome disease, that it admits of a cure with more difficulty than a carious bone, and that when destroyed, it is never recovered.” Later in 1853, Paget12 noted, “There are, I believe, no instances in which a lost portion of cartilage has been restored, or a wounded portion repaired, with new and well-formed permanent cartilage in the human subject.” Today, traumatic injuries of articular cartilage are becoming more frequent in comparably younger age groups, largely due to the increasing involvement in sports. The secondary posttraumatic changes of cartilage and bone seen in young, healthy athletes are becoming more frequent than the primary arthritic injuries seen with degeneration.
Clinical experience has demonstrated that when left untreated, these lesions usually fail to heal, and defects that involve a significant portion of the articular surface may progress to symptomatic joint degeneration. Therefore, treatment of selected isolated chondral and osteochondral defects may delay or even prevent the development of osteoarthritis.
A few studies have shown that under certain conditions articular cartilage is able to initiate a reparative process in the adult. Unfortunately, its common response is to form a repair tissue that lacks the composition, mechanical properties, structural organization, and durability that is unique to articular cartilage and vital for its normal function.8,13 Despite our knowledge, amassed over the past several hundred years, of the limited healing capacity of chondral lesions, the fundamental understanding required for the reliable treatment of articular cartilage defects has advanced little.
 Replication Process
Replication Process
The ability of the chondrocyte to replicate is an important consideration when evaluating the potential of cartilage to proliferate or repair itself. It has been shown that in the immature individual, cell replication occurs in the superficial zone beneath the tangential layer and a deeper zone adjacent to the calcified layer. However, at the time of epiphyseal closure, the chondrocyte becomes quiescent and the mitotic index decreases. At maturity, with the development of the tidemark, all evidence of mitotic activity ceases. This finding suggests that there is no compensatory mechanism to replace the cells lost to normal cell death and attrition. As such, one would expect a decrease in the number of chondrocytes with age. In reality, however, there is little or no decrease in cell count with advancing age.6 This apparent paradox has been explained by the intrinsic response and the ability of the mature chondrocyte to be able to revert to a more primitive chondroblastic state and begin the replication of its own DNA to form new cells.13
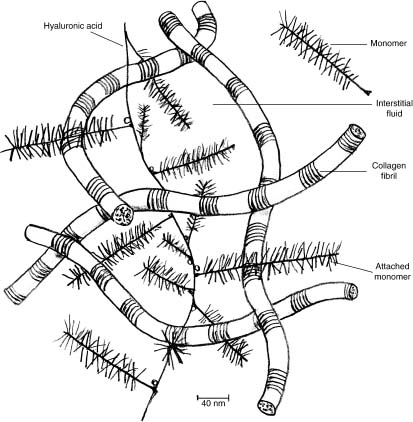
FIGURE 3–1 Collagen matrix composition consisting of collagen fibrils, hyaluronic acid, and water.
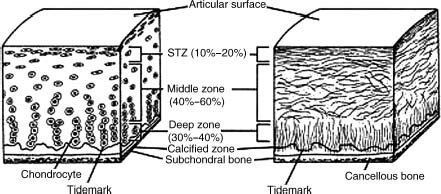
FIGURE 3–2 Chondrocyte orientation into four transitional zones, which include the tangential zone, transitional zone, radial zone, and the calcified zone. (From Buckwalter JA, Mow VC, Ratcliffe A. Restoration of injured or degenerated articular cartilage J Am Acad Orthop Surg 1994;2: 192–201, with permission of the American Academy of Orthopaedic Surgeons.)
In experimental animal models, it has been shown that following superficial lacerative articular injuries, the chondrocytes adjacent to the wound margins die or undergo apoptosis. D’Lima et al14 have shown that 34% of chondrocytes undergo apoptosis, as opposed to a baseline rate of 1%. Twenty-four hours thereafter, the adjacent cells demonstrate an increase in mitotic activity. This proliferation is associated with increased rates of synthesis of matrix components and various enzymes. Although such parameters of synthetic and degradative activity increase over the first few days following injury, they subsequently return to their preinjury levels in 1 to 2 weeks.13 As such, it appears that healing process is short-lived and does not continue to progress with time.
 Response to Injury
Response to Injury
An intrinsic response has been observed in the cells of adult articular cartilage following lacerative injury and continuous mild compression. This “intrinsic” response refers to the limited ability of mature chondrocytes to revert to their primitive chondroblastic state and replicate DNA to form new cells. Experimental studies have also shown that chondrocytes of immature and mature articular cartilage demonstrate increased rates of proteoglycan synthesis in response to a wide variety of stimuli.13 Despite numerous studies aimed at stimulating DNA synthesis in an effort to induce chondrocyte replication and increase matrix synthesis, this intrinsic reparative response is deemed clinically inadequate. Therefore, current studies have now been directed to the “extrinsic” factors, which may enhance the reparative process.
A normal response of vascularized tissues to injury is dependent on its supplying vascular network, which in turn provides the initial stages of the inflammatory response to ultimately establish a fibrin clot.9 Although cartilage is avascular, its subchondral bone has an excellent blood supply. As a result, the response of articular cartilage to a defect largely depends on the nature of the injury, that is, whether the subchondral bone has been disrupted to access the medullary vascular supply.
Superficial Articular Lesions
By lacking access to the nutrients of the subchondral bone, it follows that articular lesions which are confined within cartilage matrix fail to heal spontaneously.15–18 In contrast, articular lesions that penetrate the underlying layer of subchondral bone demonstrate a limited reparative process. In addition to accessing a plentiful vascular supply, this phenomenon has also been attributed to primitive mesenchymal cells accessing the lesion from the bone marrow and vascular networks.17,19,20 This reparative response is highly variable with respect to the quantity and quality of the tissue formed.
The repair of superficial cartilage defects depends on the response of chondrocytes at the site of injury. Numerous animal studies have demonstrated that the local response to lacerated cartilage lesions may involve some initial stages of necrosis and loss of proteoglycans18,21 that are subsequently followed by a short-lived increase in the synthesis of matrix components.18,22 In an experimental model, D’Lima et al14 have shown that the use of caspase inhibitors, which inhibit the enzymes that cause apoptosis, reduced cell death and the loss of glycosaminoglycans. This holds promise for the future, in that inhibition of apoptosis may play a role in reducing cartilage tissue damage after a traumatic injury. However, at present, there is little evidence to suggest that these lesions progress to degenerative processes within articulating joints.
Articular Lesions of Subchondral Bone
A different response is seen when the articular defect penetrates the subchondral bone. Originally, the penetration of subchondral bone was the first method used to stimulate the formation of new articular cartilage.23,24 In areas with full-thickness loss or advanced degeneration of articular cartilage, penetration of the subchondral bone (via microfracture technique, abrasion arthroplasty, or drilling) was shown to disrupt subchondral blood vessels and lead to the formation of a fibrin clot over the bony surface, which later formalizes itself into fibrocartilage (Fig. 3–3). With the surface protected from excessive loading, this fibrin clot is invaded by undifferentiated mesenchymal fibroblast-like cells.25 The subsequent release of growth factors and other proteins that influence multiple cell functions, including migration, proliferation, differentiation, and matrix synthesis, simultaneously occurs. As a result, a fibroblastic repair tissue forms that can rapidly fill the defect with cartilaginous tissue types.
Within as little as 2 weeks, primitive mesenchymal cells have assumed the rounded form of chondrocytes and have begun to produce reparative tissues consisting of chondrocyte-like cells and a large extracellular matrix of glycosaminoglycans. As determined through immunohistochemical protocols, the tissue appears to contain a significant proportion of type I and type II collagens. At 6 to 8 weeks after injury, the tissue within the chondral portion of the defects contains a relatively high proportion of the chondrocyte-like cells and a matrix composed of type I and II collagen and proteoglycans. Concurrently, cells within the bony segment of the defect form immature bone, fibrous tissue, and cartilage with a hyaline matrix. The bone formation restores the original level of subchondral bone. It rarely progresses into the chondral portion of the defect. At 6 months after injury, the subchondral bone defect has been repaired with a tissue that consists primarily of bone, but also contains some regions of fibrous tissue and hyaline cartilage.10 In contrast, however, the chondral defect is rarely completely healed.

FIGURE 3–3 Fibrocartilage filling a defect after microfracture procedure.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


