Renal Malformations
Laura S. Finn
Developmental abnormalities of the kidney produce significant rates of morbidity and mortality and are the most common cause of renal failure in children. Malformations result from a failure of normal development: “primary” anomalies are caused by disordered induction, morphogenesis, growth, and migration; “secondary” anomalies ensue from alterations superimposed on apparently normally developed kidneys. The postulated influences include maternal disease, vascular compromise, teratogens, urinary tract obstruction, and genetic mutations. Morphologic classification of malformations is traditional, but new genetic insights will lead to refined classifications that reflect pathogenetic mechanisms.
Abnormalities in migration of the entire renal unit from the pelvis lead to renal ectopia, either unilateral or bilateral. Most commonly, the kidney remains in the pelvis or may cross the midline (crossed ectopia). Fusion of the kidneys, usually at their lower poles, results in “horseshoe” kidney, with complete fusion producing a pelvic “cake” kidney. Abnormalities of ureteric branching may lead to double ureters, crossed ectopia, or bilateral displacement of the vesicoureteral insertions. These anomalies generally are asymptomatic, but they may be associated with an increased incidence of infection and, occasionally, with obstruction.
ABNORMALITIES OF RENAL MASS
Renal agenesis is complete absence of one or both kidneys and usually is associated with absence of the corresponding ureter and bladder trigone; additional anomalies of mesonephric or paramesonephric duct derivates (testes, epididymis, vas deferens, and ejaculatory duct in male patients; uterus, fallopian tubes, ovaries, and vagina in female patients) are common occurrences. Renal agenesis is seen in transgenic mice with null mutations in Wt1, Pax2, and Ret and indicates a defect in the earliest stages of nephrogenesis. The incidence of unilateral agenesis is difficult to assess because most patients are asymptomatic with only compensatory hyperplasia of the single kidney, but the condition is estimated to occur in 1 in 350 to 1,000 births. Less common and incompatible with life, bilateral agenesis occurs in approximately 1 in 5,000 births. Affected infants are stillborn or die shortly after birth from respiratory failure. Oligohydramnios results from fetal anuria and the lack of fluid, and resulting constriction produces the constellation of features termed the Potter syndrome. The infants have low-set ears, prominent infracanthal folds, a flattened beaked nose, micrognathia, creased skin, and varying positional deformities of
the limbs. Bilateral renal agenesis usually is sporadic but may be associated with hereditary renal adysplasia, a syndrome with probable autosomal dominant inheritance and variable expression, including unilateral and bilateral agenesis and dysplasia.
the limbs. Bilateral renal agenesis usually is sporadic but may be associated with hereditary renal adysplasia, a syndrome with probable autosomal dominant inheritance and variable expression, including unilateral and bilateral agenesis and dysplasia.
Hypoplasia refers to congenitally small kidneys and is an anomaly in which insufficient renal parenchyma is formed. Hypoplasia generally is sporadic, rarely familial. A decrease in the number of nephrons, frequently associated with a reduced number of renal lobes, suggests deficient ureteric bud branching or a decrease in epithelial morphogenesis. Renal hypoplasia has been associated with B-cell lymphoma-2 (BCL-2) and paired-box domain protein-2 (PAX-2) deficiencies. Hypoplasia is a rare malformation requiring morphologic confirmation. It often is confused clinically with atrophy, which can be differentiated by the presence of scarring or alterations in the architectural pattern of the kidney. Radiographic diagnosis is difficult to establish and is uncertain. Evidence of reflux or recurrent infection suggests atrophy rather than hypoplasia. Segmental renal hypoplasia (Ask-Upmark kidney) is considered to be an atrophic, rather than primary, developmental anomaly.
Bilateral renal hypoplasia ranks fourth as a cause of renal failure in children. The common form, oligomeganephronia, is an isolated and sporadic malformation characterized by extremely small kidneys that have decreased numbers of enlarged nephrons. The glomeruli are several times the norm in diameter, area, and volume, and the proximal tubules are dilated and lengthened. Oligomeganephronia occurs with a male-to-female ratio of 3:1. The major defect is in tubular concentration, leading to polyuria and dehydration. Growth retardation and moderate proteinuria are common findings when renal failure ensues. These children are good candidates for renal transplantation because the renal anomaly usually is an isolated malformation. A second, rare form of bilateral hypoplasia leading to renal failure is characterized by decreased numbers of normal nephrons.
ABNORMALITIES OF RENAL DIFFERENTIATION
Dysplasia is defined as altered differentiation of metanephric blastema. Dysplasia is seen with obstruction occurring before the completion of nephrogenesis, in association with several multiple malformation syndromes, or sporadically as an isolated event. The term is limited to kidneys showing the following characteristic histologic changes: collecting ducts lined by primitive epithelial cells with hyperchromatic nuclei; differentiated fibromuscular collars surrounding the ducts; and a loose, undifferentiated mesenchymal stroma. Metaplastic cartilage often is present in the cortex but is not an essential feature. Some metanephric differentiation always occurs, and primitive glomeruli and tubules are seen in varying numbers. Cysts, primarily involving tubules and ducts, are common findings (Fig. 324.1). Persistent fetal patterns of PAX2, BCL2, neural cell adhesion molecule (NCAM), and intercellular adhesion molecule expression (ICAM) in addition to altered growth factors and inflammatory mediators have been identified in multicystic dysplastic kidneys (MCDKs).
Renal dysplasia is associated with urinary tract malformations, particularly obstruction, in approximately 90% of cases; this finding contrasts with the lack of obstruction in hereditary and syndromal renal dysplasia. The most common of these anomalies is obstruction of the prostatic urethra in boys, a condition designated posterior urethral valves but more likely to be urethral stenosis of varying degrees of severity. When the obstruction is complete, severe bilateral dysplasia occurs. Lesser degrees of obstruction may not affect the kidney during the period of active nephrogenesis but may cause hydronephrosis or scarring later. Ureteral anomalies and malpositions, sometimes associated with ureteroceles, may cause bilateral, unilateral, or segmental dysplasia, depending on the location of the obstruction.
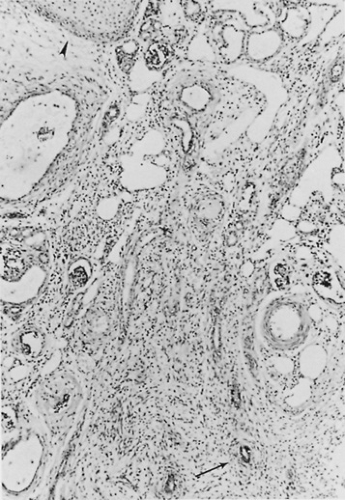 FIGURE 324.1. In this section from a cystic dysplastic kidney, primitive tubules surrounded by fibromuscular collars (arrow) are seen embedded in a loose mesenchymal stroma. Microscopic cysts and a few primitive glomeruli are present. One focus of metaplastic cartilage (arrowhead) also can be identified. (Masson trichrome, magnification 60×.) |
The most severe forms of dysplasia are MCDK and aplastic kidney, which are associated with complete ureteropelvic occlusion and ureteral atresia (Fig. 324.2). The MCDK is enlarged and has multiple cysts, has no reniform structure, and does not excrete urine. Bilateral complete disease is fatal, resulting in the Potter syndrome. Unilateral MCDK is estimated to occur in 1 in 4,300 births; most cases are sporadic. It appears to be related to obstruction occurring relatively late in nephrogenesis and usually has an inner core of more normal renal parenchyma. The aplastic-dysplastic kidney is quite small, with few cysts and little corticomedullary development. Microscopically, most of the tissue is dysplastic. This type of kidney seems to result from obstruction early in fetal development. The MCDK often is identified by prenatal ultrasonography and is the most common cause of an abdominal mass in the neonate. Lesser forms of unilateral or segmental dysplasia may remain asymptomatic for varying periods. A unilateral MCDK typically involutes, with compensatory hyperplasia of the contralateral kidney, and it may disappear, leading to a diagnosis of renal agenesis. Conservative management should include the evaluation of associated urinary tract abnormalities, particularly vesicoureteral reflux and contralateral ureteropelvic junction obstruction, which is estimated to occur in approximately 40%. In this setting, the kidneys have a higher incidence of infection. The development of hypertension may be alleviated by nephrectomy.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


