Pulmonary diseases
Chris L. Wells
Introduction
Lung disease can be classified based on its clinical characteristics. It is common to classify pulmonary diseases as obstructive, restrictive (also known as pulmonary fibrosis), vascular, infectious, disease of the pleural lining and cancers. As the population ages, it is anticipated there will be an increased burden to societies in the management of pulmonary diseases (Valente et al., 2010; Akgun et al., 2012). This chapter will briefly discuss obstructive, restrictive and infectious diseases, their clinical presentation and therapeutic interventions.
Chronic obstructive pulmonary disease (COPD) is a generic term to describe many lung pathologies that result in the trapping or retention of air upon exhalation. Emphysema and chronic bronchitis are two common obstructive diseases that affect people in the sixth decade of life. Asthma and cystic fibrosis are also considered to be obstructive lung diseases; they are typically diagnosed early in life, although asthma can develop across the lifespan.
Pulmonary fibrosis refers to diseases that cause a scarring of the lung tissue, for example interstitial pulmonary fibrosis and occupational lung diseases such as silicosis, farmer’s or coal worker’s pneumoconiosis and sarcoidosis. The result of the scarring causes a restriction or reduction of the lung’s compliance, which is the ability of the lung to expand upon inspiration.
When a lung disease results in the destruction of the massive pulmonary vascular bed, pulmonary hypertension develops within the pulmonary system. Pulmonary hypertension in the elderly can be the result of a long-standing progressive obstructive or restrictive lung disease, stenosis of the mitral valve, which causes a chronic rise in pressure in the pulmonary vascular bed, or a pulmonary embolism.
Chronic obstructive pulmonary disease
Emphysema and chronic bronchitis
The two most common diseases classified as a COPD are emphysema and chronic bronchitis. Up to 16% of people worldwide have COPD, which is believed to be significantly underestimated (American Lung Association, 2010; Akgun et al., 2012). It is projected the COPD will become the third leading cause of death by 2020 (Valente et al., 2010).
Emphysema is defined as irreversible anatomical enlargement of the airspaces that are distal to the terminal bronchioles (Fig. 45.1). There is destruction of the acini, which are the functional units of the lung where gas exchange occurs in individuals without fibrosis. Emphysema can be classified based on the location of the anatomical disruption. Centrilobular emphysema is the type of emphysema most commonly associated with smoking and involves the enlargement and destruction of the first- and second-order respiratory bronchioles with the alveoli remaining intact. It most commonly affects the upper lobes and results in a mismatch between ventilation and perfusion. Panacinar emphysema is commonly found in the elderly and in patients who have a genetic form of emphysema called α1–antitrypsin deficiency. This form of emphysema affects all of the respiratory bronchioles in a uniform pattern. Paraseptal emphysema involves the peripheral secondary lobules and is not typically associated with progressive end-stage disease but with an increased risk and incidence of pneumothorax. Finally, paracicatricial emphysema is characterized by irregular enlargements of the acini with fibrosis, usually adjacent to a previous pulmonary lesion (Hogg, 2004).
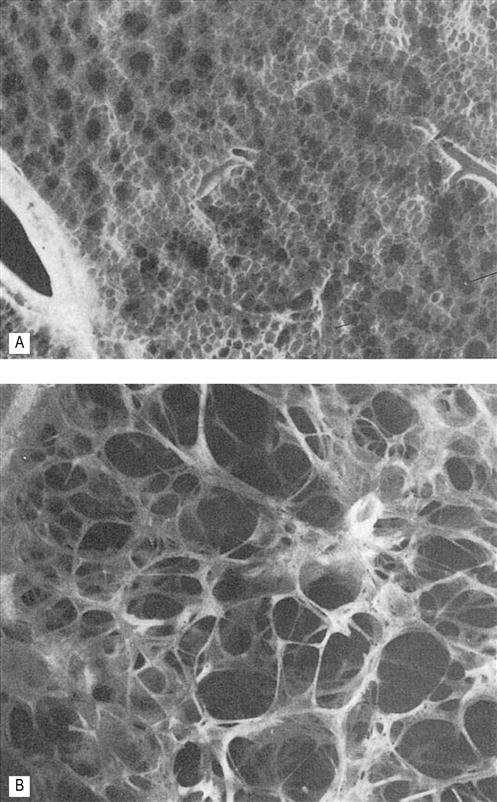
Emphysema is the second most common of the obstructive diseases. There is a definite increase in the incidence of emphysema in the fifth decade and a continued increase into the seventh decade with the prevalence exceeding 100 cases per thousand (Akgun et al., 2012). Because the lungs have a vast amount of surface area to allow for sufficient gas exchange, many individuals will be asymptomatic in the early stages of the disease unless the activity level is at a high intensity; under such conditions, emphysema may contribute to the fatigue and shortness of breath that individuals relate to deconditioning, the aging process, or other comorbidities (Valente et al., 2010). The level of disability or functional limitation is dependent on the extent of lung destruction, not the type of emphysema, age, number or severity of comorbidities and level of function (Vorrink et al., 2011; Messer et al., 2012).
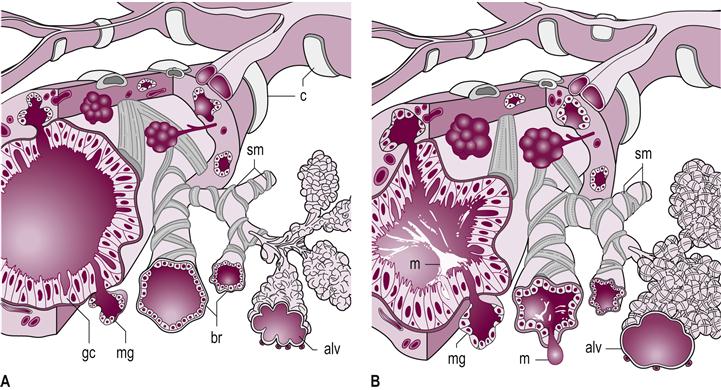
Chronic bronchitis is a leading COPD disease and is twice as common in women, with a reported prevalence of 57.7 per thousand (Valente et al., 2010). Chronic bronchitis and emphysema are treated as one long continuum, with chronic bronchitis clinically presenting with a persistent productive cough that produces sputum for more than 3 months per year for at least two consecutive years in the absence of another definable medical cause for the sputum production, such as pneumonia. The disease is associated with hyperplastic glands and an increase in goblet cells of the epithelial lining. There is a marked decrease in the ratio of goblet cells to ciliated cells, leading to hypersecretion of mucus, which overwhelms the mucociliary clearance (Fig. 45.2). The end result is overproduction and retention of sputum, which causes airway obstruction, inflammation of the respiratory bronchioles, narrowing or occlusion of the small airways from mucus plugs and hypertrophy of the smooth muscle, resulting in an increased risk of pulmonary infections (American Lung Association, 2010).
Contributing factors
Several contributing factors have been linked to emphysema and chronic bronchitis, the most common of which is cigarette smoking. Smoking increases the aggregation of neutrophils and alveolar macrophages, which begin the immune response to rid the body of foreign materials. One theory suggests that emphysema is the result of an imbalance between the protective antiprotease enzymes that protect the delicate structures of the lungs and the protease enzymes that lyse or break down tissue. This imbalance leads to the loss of the elastic recoil of the lung architecture. The small airways depend upon the adjacent elastic tissue of the parenchyma to recoil and assist with expiration and to provide airway stability to allow for effective inspiration. Another theory currently under examination is the role that smoking plays in the observed elevated rate of apoptosis or cell death of the alveolar cells, followed by the failure to repair the structures to a functional state. There is also an increase in platelet and neutrophil aggregation, which destroys the small capillary beds. This leads to a decrease in the gas exchange function of the lungs and pulmonary hypertension (Higenbottam, 2005; Provinciali et al., 2011).
Exposure to air pollutants and occupational factors is also associated with an increased incidence of emphysema and chronic bronchitis. There is an increased incidence in the development and progression of COPD as the number of respiratory infections increases. The increased frequency and dose exposure to systemic steroids, for example prednisone, has also been linked to the progression of COPD.
Finally, age and genetic factors have also been linked to the development of COPD. With the increase in the age of the population there has been a significant rise in the prevalence of COPD (Akgun et al., 2012; Rycroft et al., 2012). There is a genetic link with a predisposition to emphysema and chronic bronchitis, with a reported association with a decrease in the level of alpha-1-antitrypsin (Akgun et al., 2012). This enzyme is a protease inhibitor which protects tissue from enzymes of inflammatory cells, such as neutrophil elastase. With the reduction in alpha-1-antitrypsin there is an increased rate of cellular breakdown. Within the lungs this leads to emphysema.
Clinical manifestation
Emphysema and chronic bronchitis can exist without evidence of clinically significant obstruction or functional limitations. However, by the time that the patient presents to the healthcare provider with symptoms, extensive irreversible lung damage is present. COPD results in a limitation of airflow. The lumen size of the bronchioles is decreased because of smooth muscle proliferation and contraction, and because of bronchial edema resulting from inflammation. With the loss of lung parenchyma, there is a reduction in the elastic recoil of the airways, leading to dilatation of the distal airways and early airway closure. The end result of these changes in structure within the lungs is an increase in the ease with which air can enter the lungs during inspiration, which is referred to as an increase in compliance. Unfortunately, the changes also result in the closure or collapse of the fragile airways upon exhalation, leading to air becoming trapped in the distal respiratory bronchioles and acini. This presents clinically as hyperinflation of the lung. The hyperinflation causes shortening of the inspiratory muscles and a flattening of the diaphragm. This leads to compensatory changes in the chest wall called barrel chest deformity, which is an increase in the anterior–posterior dimension and rib angle. These musculoskeletal changes lead to a decline in the mechanical effectiveness of the diaphragm and other respiratory muscles to support the increased demands of ventilation.
The clinical presentation of patients with COPD has an insidious onset and goes undiagnosed for many individuals. Common symptoms are nonspecific and include chronic cough, dyspnea, possible sputum production and high-pitched wheezing. With the progression of the disease there is an increased work of breathing and a reduction in activity level (Vorrink et al., 2011). Upon auscultation, there are diminished normal breath sounds and high-pitched wheezing, particularly associated with exertion. There is an elongated expiratory phase because the patient tries to slow down the change in airway pressure during expiration to minimize the degree of air trapping or obstruction. Accessory respiratory muscles are commonly hypertrophied and there is a decrease in the excursion of the diaphragm as the lungs hyperinflate. On percussing over the intercostal spaces, there is hyper-resonance. With exertion, there is a marked increase in muscle recruitment, both for inspiration and expiration. Shortness of breath is the leading cause of exercise intolerance.
Patients who have a mismatch between ventilation and perfusion have the clinical presentation of desaturation because of the disruption of gas exchange. There may be areas of the lung where there is good blood flow through the pulmonary capillaries but poor ventilation. There may also be areas with an increase in the dead space in patients with COPD, which means that there is sufficient ventilation occurring in areas of the lungs where the capillary bed has been destroyed or pruned. This mismatch between ventilation and perfusion leads to hypoxia and the retention of carbon dioxide.
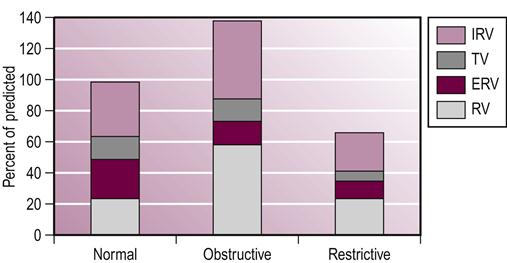
Examination of pulmonary function tests in patients with COPD reveals a classic pattern. Patients have a marked reduction in the ability to expel air rapidly, measured by the forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC). The decline in FEV1 is associated with the degree of dyspnea or shortness of breath. Exercise limitations because of dyspnea are associated with a FEV1 of less than 50% of the predicted value for age, height and weight (Barnes & Fromer, 2011). When the patient is dyspneic at rest, the FEV1 may be as low as 25% of the predicted value. There is also a marked increase in total lung capacity and residual volume, which clearly represents the increased compliance of the lung and the degree of air trapping or obstruction (see Fig. 45.3). It is recommended by the American Association of Respiratory Care and COPD Foundation that questionnaires should be commonly used on all adult patients to identify risk factors for COPD and simple hand-held spirometer or peak flow meters to motor airflow. For those individuals with risk factors and low expiratory flow a high-quality spirometry testing should be completed (Barnes & Fromer, 2011).
Although the majority of patients with COPD have mixed features of both emphysema and chronic bronchitis, there are certain clinical signs and symptoms that are associated more with emphysema than with chronic bronchitis. With emphysema, there is a long history of dyspnea on exertion and little sputum production. These patients favor a posture of forward trunk flexion; this is to fixate their upper extremities so that accessory muscle recruitment is increased and the influence of gravity is decreased. The patient with emphysema is more likely to practice pursed lip breathing or grunt during expiration to keep the airways open. The patient will present with an elevated minute ventilation (respiratory rate×tidal volume), which aids in maintaining a sufficient arterial oxygen concentration at least through the early to mid-stages of the disease. In addition, a patient who suffers predominantly from emphysema will have an underweight to cachectic appearance (Hogg, 2004; American Lung Association, 2010).
In contrast, a patient who suffers predominantly from chronic bronchitis usually presents with a long history of a chronic and productive cough. Initially, the productive cough may occur only during the winter months; however, as the disease progresses in duration, frequency and severity, there is excessive sputum production and mucopurulent infections. By the time the patient experiences exertional dyspnea, there is a severe degree of airway obstruction. These patients have a tendency to be overweight and cyanotic, with a lower minute ventilation than patients with emphysema (Hogg, 2004).
As these diseases progress to end-stage, there will be further declines in lung function. The destruction of the respiratory bronchioles and acini lead to additional difficulties with proper ventilation, an increased airway resistance and a significant increase in the work of breathing. The disruption of the capillary bed within the lung causes a rise in pulmonary pressure and places a strain on the right ventricle. Over time, the patient will develop cor pulmonale, or right heart failure, which is associated with peripheral pitting edema, ascites and enlargement of the liver, jugular vein distension and anorexia.
Therapeutic intervention
Smoking cessation is instrumental in the care of patients with COPD. Cessation leads to a decrease in the rate of loss of FEV1. Importantly, cessation also means the avoidance of people or places where there is the risk of exposure to second-hand smoke. Behavioral modification training should also focus on weight management and healthy eating, developing coping strategies to minimize anxiety attacks, controlling responses to stress and learning breathing strategies to control dyspnea.
Beyond behavioral modifications there are many pharmacological options to assist in the management of the disease and the associated symptoms. Short- and long-acting β2-agonists or bronchodilators such as albuterol can be used to minimize bronchospasm and decrease wheezing and airway resistance. Anticholinergic drugs, such as atrovent, can block bronchoconstriction; xanthine-derived medications, such as theophylline, also produce bronchodilation and accelerate the mucociliary transport system and limit the inflammatory response. Corticosteroids, such as prednisone or flovent, are used for their anti-inflammatory benefits. Considerations need to be made when prescribing these medications to older adults due to pharmacokinetic and pharmacodynamic differences due to effects of aging. It is critical for patients with lung disease to receive a flu shot annually to decrease the risk of infection. When a patient has a history of recurrent infections, antibiotics play a critical role not only in the treatment of a recurrent infection but also as part of prophylactic care (Valente et al., 2010; Abbatecola et al., 2011; Jen et al., 2012).
These patients may also benefit from airway clearance techniques, including postural drainage, percussion and assistive breathing techniques, mobilization, or the use of an oscillating device to mobilize secretions. Finally, supplemental oxygen is used to correct hypoxemia and minimize secondary pulmonary hypertension. Oxygen therapy has been shown to reduce the level of dyspnea, decrease pulmonary hypertension, reduce the incidence of cardiac arrhythmias and improve quality and quantity of life. In the patient who presents with hypercapnia and respiratory insufficiency, bilevel positive airway pressure (BiPAP) ventilation has become recognized as an effective device in the management of patients with progressive disease. The ventilator provides positive airway pressure to decrease the work of inspiration and minimize the air trapping, which can reduce the retention of carbon dioxide.
Pulmonary rehabilitation has become a widely accepted intervention in the care of patients with COPD, with the ultimate goal of improving quality of life. The therapy program should consist of a comprehensive educational program to address such issues as nutrition, weight management, pathology and medical management, including the proper use of medications, work simplification and coping strategies. The program should stress and progress aerobic tolerance and include weight training to improve the muscular strength and endurance of the upper body; this will increase the effectiveness of the accessory respiratory and antigravity muscles in maximizing breathing and promoting functional mobility. The exercises should be functional and weight-bearing in nature to aid in the management of osteopenia and osteoporosis, which are very common in this patient population. Oxygen saturation should be monitored closely and supplemental oxygen adjusted to provide sufficient perfusion to support aerobic training. With training, the majority of patients with COPD will improve their exercise capacity, and note a decrease in the perception of dyspnea, an increase in self-control and a marked improvement in quality of life (Barnes & Fromer, 2011; Burtin et al., 2011; Nagarajan et al., 2011).
There are surgical options for the treatment of emphysema and chronic bronchitis. In the presence of a large bulla, which is a large airspace that is no longer contributing to gas exchange and is compressing adjacent tissue, surgical resection of this tissue (bullectomy) can be performed. Volume reduction surgery is an option for patients with emphysema, in which about 20% of dysfunctional lung tissue is surgically removed to decrease hyperinflation and improve ventilation and perfusion. Finally, lung transplantation has become a viable option for patients with end-stage COPD who have maximized medical therapy (Nathan et al., 2004).
The prognosis for patients with COPD varies depending upon the degree of obstruction, the presence of hypercapnia, the level of hypoxemia, functional mobility, body mass index, the recurrence of infections and country that the individual resides in; the 1-year mortality rate for all disease levels was reported at 27.7% and 5.1% in Canada and Sweden respectively (Rycroft et al., 2012). It is generally accepted that a FEV1 of less than 25% is associated with a 50% mortality rate within 2 years. In chronic bronchitis, the prognosis is dependent upon age, smoking and the degree of airway obstruction. The 10-year survival rates for individuals with COPD over the age of 50 years is less than 30%, approximately 50% and 63% stratified by severe, moderate and mild disease (Shavelle et al., 2009).
Pulmonary fibrosis
Pulmonary fibrosis is the name for the hundreds of pulmonary pathologies that result in a restriction of the lungs. The ability of the patient to increase the volume of air in the lungs, or lung compliance, diminishes as the disease progresses; the compliance of the chest wall also decreases because of the decrease in range of motion of the chest wall, ribs and thoracic spine. Pulmonary fibrosis can be caused by autoimmune diseases, such as rheumatoid arthritis, lupus and scleroderma, or can result from occupational exposure, such as farmer’s lung, silicosis and black lung. Pulmonary trauma, fat embolism and infection are associated with the development of acute respiratory distress syndrome, which for a small portion of individuals may result in pulmonary fibrosis. Other diseases, for example interstitial pulmonary fibrosis, are idiopathic in nature and for others the disease has been linked to the adverse effects of medications.
Occupational disease
There is a subset of pulmonary interstitial disorders that result from the inhalation of inorganic dusts (pneumoconioses), organic particles (hypersensitivity pneumoconioses) and industrial gases, fumes and smoke. Fifteen percent of adult onset asthma has been linked to occupational exposure. These occupational lung diseases are associated with a chronic inflammatory process and fibrogenesis that leads to destruction of the alveolar capillary membrane. The end result is arterial hypoxemia (Noble et al., 2010; Schmidt & Flaherty, 2011; Wynn, 2011).
Pneumoconioses involves the permanent deposition of inorganic material (coal, asbestos, silica, beryllium, etc.) within the pulmonary system. The risk of developing pulmonary fibrosis is related to the duration and intensity of exposure and the size and water solubility of the particles. There is a long latency between exposure and disease, sometimes as long as 20–40 years, which may place the onset of disease in the fifth to seventh decades of life.
If the inorganic materials are able to get beyond the ciliary structures of the nasal passage and mucociliary blanket, they may cause an inflammatory process within the air spaces and interstitium, resulting in lung injury. Hyperplasia and proliferation of pulmonary epithelial cells characterize the immune response, which is accompanied by fibroblastic proliferation and collagen and protein deposition.
Hypersensitivity pneumonitis, or external allergic alveolitis, is an immunologically mediated disease that is typically associated with sensitivity from repeated exposure to an antigen. Further exposure results in an inflammatory response that involves the distal airways and alveoli. There are numerous agents that can be the impetus for developing hypersensitivity pneumonitis; these include moldy hay or grains, fungi from water reservoirs, bird serum, feathers and excreta, mining dust and pharmacological products such as gold, amiodarone and minocycline. The inflammatory response persists beyond the exposure time and leads to permanent lung damage. There is infiltration of macrophages and lymphocytes, and epithelioid granuloma formation, which eventually leads to obliteration of the bronchioles because of scarring. If the disease progresses into the chronic phase, the granulomas disappear and are replaced by fibrotic tissue formation and destruction of the architecture of the lung (Harari & Caminati, 2010).
Acute respiratory distress syndrome
Acute respiratory distress syndrome (ARDS) is an acute lung injury that results in bilateral pulmonary infiltrates, severe refractory oxygenation, noncardiac pulmonary edema and an increase in lung stiffness (i.e. a decrease in compliance). It has been suggested that ARDS is the severest form of pulmonary edema, in which diffuse alveolar involvement proceeds to promote further injury. ARDS is associated with high mortality and morbidity rates; 60-day mortality rates have decreased but still remain at 22–35%. Mortality is associated with severity of hypoxia, age over 60 years, liver or renal dysfunction or failure, exposure to tobacco smoke and alcohol use (Mann & Early, 2012; Matthay et al., 2012).
The most common cause of ARDS is a bacterial or viral infection but it can also be the result of sepsis from a nonpulmonary infection, aspiration, major trauma, blood transfusions, or pancreatitis. Diffuse alveolar disease is the hallmark picture of ARDS. Whatever the source, the injury is either to the alveolar membrane or vascular endothelium. This leads to increased permeability and a shift of protein-rich exudate into the alveoli. Subsequently there is pulmonary edema and hypoxemia (Mann & Early, 2012; Matthay et al., 2012).
This heterogeneous disorder changes over time. The initial phase (exudate phase) is characterized by pulmonary edema, hemorrhage and hyaline membrane formation. Clinically, there is a rapid onset of respiratory failure that is refractory to supplemental oxygen. The second phase involves cellular proliferation, with an elevation in the number of neutrophils and other inflammatory cells. This phase is characterized by diffuse alveolar disease (DAD); this is associated with cellular necrosis, epithelial hyperplasia and further inflammation, which leads to destruction of the delicate structures of the lung. The third phase is fibroproliferation, which is the result of chronic inflammation whereby injured lung tissue is replaced with fibrotic tissue. Beyond the destruction of terminal bronchioles and alveoli, there is also obliteration of the pulmonary capillaries, leading to pulmonary hypertension and, eventually, right heart failure (Mann & Early, 2012).
Treatment includes identifying and treating the underlying cause, mechanical ventilation, steroids, sedation and paralytics to reduce oxygen consumption needs, and extracorporeal membrane oxygenation (ECMO). Prescription of mechanical ventilation is still under debate, with most agreeing that the use of positive end-expiratory pressure is critical for gas exchange and to reduce further lung injury from positive mechanical ventilation; there are reports that the use of lower tidal volumes with elevated respiratory rates is also beneficial (Matthay et al., 2012; York & Kane, 2012).
Idiopathic pulmonary fibrosis
The onset of idiopathic pulmonary fibrosis (IPF) occurs in mid to late life with high mortality and morbidity rates. There is variability in the progressive rate of IPF and has a 20–30% 5-year survival rate (Harari & Caminati, 2010; Noble et al., 2010). There is an increased incidence with males and increasing age. Risk factors also include a history of smoking (Noble et al., 2010). Idiopathic pulmonary fibrosis can be associated with usual or desquamative interstitial pneumonia. Usual interstitial pneumonia (UIP) is characterized by patchy, non-uniform and variable destruction of interstitial tissue. There is a minimal inflammatory component to this disease, involving collagen deposition that thickens the alveolar septum. Desquamative interstitial pneumonia (DIP) is another form of IPF that presents with little fibrosis but a significant inflammatory response, with an accumulation of alveolar macrophages within the alveolar spaces and interstitium. The initial injury appears to damage the alveolar and epithelial cells, causing inflammatory cells to release cytokines, tumor necrosis factor and platelet-derived growth factor. These inflammatory chemicals result in smooth muscle proliferation, degradation of the alveoli and the proliferation of fibroblasts and an increase in collagen deposition. With IPF there appears to be abnormal responses during the inflammatory and fibroblastic proliferation phase of healing that leads to advanced scarring and alveolar destruction (Noble et al., 2010; Wynn, 2011).
Clinical manifestation
Despite the range of etiologies that result in pulmonary fibrosis, patients present with a similar clinical picture of a variable rate of progressive decline, exertional dyspnea, a nonproductive cough that worsens with exertion and severe cyanosis. Along with severe dyspnea, patients generally experience severe desaturation with exertion. There is a decrease in normal breath sounds and the development of rales and clubbing of the nail beds. The breathing pattern is typically shallow with an elevated rate and there is a reduction of rib cage mobility, leading to a marked increase in the work of breathing. Anorexia, malaise and muscle weakness are also common clinical signs. Finally, pulmonary fibrosis is commonly associated with pulmonary hypertension and right heart failure (Markovitz & Cooper, 2010).
Conventional chest radiography reveals diffuse infiltrates, and honeycombing develops in the later stages of pulmonary fibrosis. When pulmonary function tests are examined, there is a decrease in lung volume, especially vital capacity (VC) and total lung capacity (TLC), a decrease in the gas exchange ability of the respiratory system (diffusion capacity; measured using the diffusing capacity of the lung for carbon monoxide [DLCO] test) and a decreased pulmonary compliance with a normal FEV1–FVC ratio (see Fig. 45.3). A ventilation–perfusion mismatch occurs as ventilation declines and is associated with severe hypoxia. It has also been documented that there is skeletal muscle dysfunction (e.g. a reduction in type 2 muscle fibers) in patients with pulmonary fibrosis which is linked to disuse atrophy, adverse effects of medication therapy and the untoward effects of a prolonged elevated level of inflammatory markers (Markovitz & Cooper, 2010).
Therapeutic intervention
The best defense against pneumoconiosis is prevention; this is achieved with the use of proper respiratory filter devices and appropriate ventilation in the work area. Management includes the use of corticosteroids to minimize the inflammatory response and monitoring the progression of the disease with radiological studies, pulmonary function tests and exercise testing. Medications that inhibit the immune response are also utilized in the treatment of IPF. Cyclophosphamide impairs the function of neutrophils, eventually decreasing fibroblast and collagen proliferation. Azathioprine and cyclosporin (ciclosporin) suppress the production and maturation of T and B cells involved in the immune response. With the progression of the disease, medical care may include the use of supplemental oxygen and prostacyclin drugs for the treatment of right heart failure resulting from pulmonary hypertension. Non-invasive mechanical ventilatory support may be helpful to decrease the work of breathing, improve oxygen delivery and allow for rest periods. In the presence of isolated pulmonary fibrosis, lung transplantation should be considered on a case-by-case basis (Harari & Caminati, 2010).
Pulmonary rehabilitation can also be beneficial for patients with pulmonary fibrosis with improvement in 6-minute walk distance, decrease in dyspnea levels and improvement in quality of life. Once again, the ultimate goal is to improve the quality of life. An educational and exercise program, similar to that described in the COPD section, should be provided for this patient population but the clinician should expect the rehabilitation progress to be much slower than with COPD patients. It is important that the exercise program be tailored to the individual patient with pulmonary fibrosis (Markovitz & Cooper, 2010). The program should include stretching exercises to maintain chest wall mobility and, in the presence of pulmonary hypertension, interval exercises. Prescribed rest times are essential to decrease the strain on the right ventricle. The rehabilitation prescription should also include aerobic training, functional muscle strength and endurance training. These patients typically require high levels of supplemental oxygen to prevent severe hypoxia so the rehabilitation clinician will need to work closely with the pulmonologist and respiratory therapist to prescribe the proper supplemental oxygen delivery systems and oxygen prescriptions. The disease progression is generally aggressive, so work simplification training is also valuable and the program should be focused on maintaining functional mobility.
The prognosis is generally poor for patients with pulmonary fibrosis because of refractory hypoxemia, right heart failure and the increased risk of bronchogenic carcinoma in patients who smoke and those with occupational exposure pulmonary fibrosis. In ARDS, the mortality rate is as high as 60%, but has been decreasing to 22–35%, with a higher death rate in older patients (Matthay et al., 2012). In general, the mean survival time in cases of pulmonary fibrosis is 2–5 years but this will vary based on the aggressiveness and type of disease, duration of symptoms and response to therapy (Harari & Caminati, 2010; Noble et al., 2010).
Pulmonary hypertension
Secondary pulmonary hypertension can be the sequela of a congenital heart defect, collagen vascular disease, lung disease, COPD and pulmonary fibrosis, hypoxia, thromboembolic disease and left heart failure resulting from cardiomyopathy and valvular disease. As pulmonary disease progresses to the point where the pulmonary capillary bed becomes affected, pulmonary pressure begins to rise. Pulmonary hypertension can be defined as a mean pulmonary arterial pressure that is greater than 25 mmHg at rest and greater than 30 mmHg during exercise.
A significant amount of the lung parenchyma must be involved to cause pulmonary hypertension because the reserve capacity of the lungs is so vast. As the intrinsic obstructive lung disease progresses, the disruption of capillary beds and destruction of the gas exchange area of the parenchymal tissue leads to hypoxia and vasoconstriction, producing pulmonary hypertension. Precapillary arteries and arterioles also become less distensible and constricted. With pulmonary fibrosis, for example in collagen vascular disease, the scarring of the airways and capillaries causes a decrease in compliance and arterial hypertension. The consequences of abnormal and chronic vasoconstriction include intimal proliferation, smooth muscle hypertrophy and changes in the endothelium that lead to a decrease in the diameter of the arterial lumen and vascular remodeling. With end-stage heart disease there is an increased production of endothelin1 and thromboxane A2 and reduction in nitric oxide leading to smooth muscle cell hypertrophy and hyperplasia. These changes, along with venous congestion, lead to elevation in pulmonary arterial pressures (Mikus et al., 2011; Akgun et al., 2012).
If the pulmonary pressure is not relieved, the pulmonary vascular system becomes less distensible and blood is shunted to the larger vessels, which causes a ventilation–perfusion mismatch. To compensate for the elevated pulmonary vascular resistance and to maintain cardiac output, the right ventricle hypertrophies. Over time, the myocardium dilates and is unable to maintain efficient blood flow through the lungs for gas exchange, leading to heart failure (Mikus et al., 2011).
Clinical manifestation
The progression of dyspnea and the early onset of fatigue are typically the first symptoms of pulmonary hypertension, although many patients associate this with aging and deconditioning. Patients may begin to complain of presyncopal symptoms or may experience syncope. Chest pain, muscle fatigue, hypoxemia and hemoptysis are other common symptoms related to pulmonary hypertension. As the patient develops cor pulmonale, the signs and symptoms of right heart failure become present, which include jugular vein distension, peripheral edema and hepatic congestion. (See Chapter 42 for descriptions of the signs and symptoms of heart failure.)
Upon examination, the right ventricle may be palpable in the lower left sternal or subxiphoid area and abnormal heart sounds are present, including S4 gallop and a split S2 sound. As the disease progresses, S3 gallop can be heard, indicating advanced right heart failure. Abnormal valvular heart sounds may also be audible, including a systolic ejection click and tricuspid murmur. The electrocardiogram (ECG) is consistent with right ventricular hypertrophy and changes in the T wave. As the disease progresses, there will be clear signs of right heart failure, in most cases including jugular vein distension, hepatic congestion, peripheral edema, ascites and systemic hypotension (Higenbottam, 2005).
Therapeutic intervention
The treatment of pulmonary hypertension involves treating the primary cause of it. Drugs to decrease strain on the right side of the heart, such as digitalis and diuretics, and supplemental oxygen therapy to treat the hypoxemia may be effective. Continuous intravenous prostacyclins, for example Flolan and Iloprost, may decrease pulmonary hypertension by vasodilatation when infused into the pulmonary arterial system. The most common positive effects of the intravenous use of prostacyclins are an improvement in exercise tolerance and a decrease in symptoms experienced at rest and with exertion. Endothelin receptor antagonists, such as bosentan, reverse the effects of endothelin, and sildenafil (Viagra) and tadalafil (Cialis) are vasodilators also used in the management of pulmonary hypertension. Anticoagulation medications may be used to decrease the risk of thromboembolic events because of polycythemia, which may develop as a compensatory mechanism to offset hypoxemia. In the case of heart failure, ventricular assist devices may be used to manage, and in some cases reverse, the levels of pulmonary hypertension (Mikus et al., 2011).
Rehabilitation for patients with pulmonary hypertension typically focuses on functional mobility. It is also important to review job simplification and energy conservation in these patients. Patients typically tolerate an interval aerobic program, particularly a walking program. Exercises that isolate muscle groups, such as cycling, are usually less well tolerated because of local muscle fatigue. It is important that the therapist prescribe an exercise intensity that is sufficient for the patient to experience the benefits of exercise without causing abnormal responses to exertion. These patients should be monitored closely for signs of chest discomfort, lightheadedness or excessive fatigue. The therapist should also educate the patient about the adverse signs and symptoms that indicate distress and progression of the disease. It is also vital that the therapist works directly with the physician to establish safe parameters for functional mobility activities (see Box 39.4 and Tables 39.4–39.7 for guidelines).
Pulmonary embolism
Pulmonary embolism is closely linked to the presence of blood clots or thrombi in the peripheral venous system, known as deep vein thrombosis (DVT). Pulmonary embolism is the third leading cause of cardiovascular deaths and has a 10 per 1000 cases incidence in older adults versus 1 per 1000 cases in young adults (Geersing et al., 2012). Typically, the source of the embolism originates from a DVT in the upper legs or pelvis. Small emboli may present little compromise to a healthy individual but may cause severe respiratory failure in an older individual with a reduced reserve of the cardiopulmonary systems. In the elderly, there are several risk factors that should be part of the clinician’s screening process, including previous DVT or pulmonary embolism, surgery, malignancy, hormonal therapy, obesity, venous stasis, immobility, cerebrovascular accident (CVA), recent trauma and heart failure (Geersing et al., 2012; McLenon, 2012). In the older adult PE are underdiagnosed, with up to 40% of PE found at autopsy in older adults (Geersing et al., 2012).
Clinical manifestation
The most pronounced clinical presentation in cases of pulmonary embolism includes unexplained dyspnea of rapid onset, pleuritic chest pain and hypotension with no obvious cause. The presence of hemoptysis indicates pulmonary hemorrhage or infarction (McLenon, 2012).
During evaluation, it is important to develop a differential diagnostic list and proceed with testing to enable a clinical diagnosis to be formulated. The differential diagnosis may include the following conditions: acute myocardial infarction (MI), asthma, pneumothorax, congestive heart failure (CHF), acute pulmonary edema, pleurisy, pericarditis, musculoskeletal trauma to the chest wall, sepsis, tamponade and aortic dissection. Risk factors associated with PE and prognosis include age>80, male gender, history of cancer, heart failure, chronic lung disease, previous DVT or PE, renal or liver disease, recent trauma or surgery (Geersing et al., 2012).
Upon physical examination there may be a low-grade fever, cyanosis, tachycardia, jugular vein distension, tachypnea and hypotension. Upon auscultation, there may be a pleural rub and a split of the S2 heart sound may be heard over the pulmonic valve. The degree of respiratory compromise is dependent on the size of the pulmonary embolism and the preexisting cardiopulmonary reserves. An echocardiogram may be suggestive of right heart strain or ischemia and the ECG may demonstrate T-wave inversion.
Clinical intervention
The key to appropriate medical care is the identification of patients who are at high risk and implementation of effective prophylactic treatment. Treatment includes prevention such as early mobilization and the use of graduated compression devices and TED stockings. The use of intermittent pneumatic compression stockings provides peripheral pumping to encourage venous return and reduce venous stasis. Many patients will be prescribed anticoagulants for the prevention and treatment of DVT formation. In patients who cannot take anticoagulants, an inferior vena cava filter may be placed to decrease the risk of a pulmonary embolism occurring from a lower extremity or pelvic thrombus. Thrombolytic therapy has also been used successfully to break down the DVT or pulmonary embolism, but is associated with a risk of hemorrhage (McLenon, 2012). Finally, pulmonary endarterectomy has become a viable surgical option to remove the emboli from the pulmonary artery or arteries. It is performed through a sternotomy and can be complicated by persistent hypoxia and prolonged mechanical ventilation as well as management of heart failure.
The prognosis for a patient suffering from a pulmonary embolism is dependent upon the size of the pulmonary embolism, the underlying compromise of the cardiopulmonary system and the promptness of medical care. Mortality rate for PE has been reported to be as high as 30% if untreated and 80% of unexplained hospital deaths were related to undiagnosed PE. There is a 10% 1-year mortality for individuals in the first year after diagnosis of PE. Mortality can be as high as 40% within 5 years in individuals who stopped anticoagulation therapy (Geersing et al., 2012; Ouellette & Mosenifar, 2013).
Pulmonary infections
Pneumonia
The pulmonary system has two primary mechanisms to manage the presence of foreign matter that may precipitate a pulmonary infection. The upper airway warms and humidifies the air and the mucociliary cells aid in the entrapment of particles in this conductive system of the lungs. If particles enter the lung, there is an immune response that attacks the foreign material and removes it. When one or both of these mechanisms is impaired, there is an increased risk of developing a pneumonia, which is defined as an acute inflammation of the lungs, causing the small bronchioles and alveoli to become plugged with fibrous exudate.
Pneumonias can be classified based on several parameters: (i) by the etiology underlying the infection, including bacterial, viral and fungal sources; (ii) as typical or atypical, based on the incidence of the infection in a given population or location; and (iii) by the site in which the infection occurs, with acquired pneumonias referring to infections obtained in the community and nosocomial pneumonias defined as infections that occur during the hospitalization of the patient. With the increase in admissions to such facilities as long-term care or nursing homes, acquired infections may be subdivided into community-acquired and institutionally-acquired pneumonia. However the infection is classified, there are common risk factors that contribute to the susceptibility of developing pulmonary infections (see Box 45.1) Acquired pneumonias account for 650 000 annual hospital admissions and has a prevalence of 14 out of 1000 cases. This number is doubled for nursing home residents where aspiration-related pneumonias have a 2–4 times incident rate (Akgun et al., 2012).





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


