Fig. 17.1
Blunt impact suppresses cartilage ATP production. Explant cartilage was harvested for ATP assay 24 or 48 h postimpact (7 J/cm2). Cartilage was harvested from control (non-impacted) explants and from impacted explants from the impact site itself, from a site immediately adjacent to the impact, or from a remote site several mm distant from the impact. One-way analysis of variance indicated that at 24 and 48 h postimpact, ATP in the impacted and adjacent cartilage was significantly lower than in cartilage from control explants. Remote cartilage ATP content was significantly lower than control at 24 h, but not at 48 h postimpact (*p < 0.001). Columns and error bars indicate means and standard deviations (n = 4)
Oxidants produced by chondrocytes in response to cartilage deformation are mitochondrial in origin [5, 39]. Metabolic inhibition experiments revealed that O2 •− radicals are released from the mitochondrial electron transport chain in a strain-dependent fashion. When cartilage in the center of osteochondral explants (15 mm × 15 mm) is compressed with varying stresses (0.1–1 MPa) (imparted by an 8 mm diameter nonporous steel platen), the number of chondrocytes producing detectable O2 •− increased linearly from 10 to 60 % strain (r 2 = 0.87) (Fig. 17.2) [4]. Strains below 40 % were well tolerated (<15 % death), but the death rate increased sharply as strains rose above 40 %. O2 •− release and cell death were suppressed by rotenone, an electron transport inhibitor, and by cytochalasin B and nocodazole, which inhibit the polymerization of actin and tubulin respectively [40]. Additional work revealed O2 •− overproduction and a chondrocyte mortality rate of ~60 % in explant cartilage subjected to a single blunt impact injury [6]. It is noteworthy that death in this model occurred mainly near matrix cracks where the greatest matrix strains occurred during the impact load. This effect was also mitigated by a variety of drugs targeting strain-induced O2 •− production including antioxidants, rotenone, inhibitors of cytoskeleton polymerization, and inhibitors of cell-ECM adhesion [41].
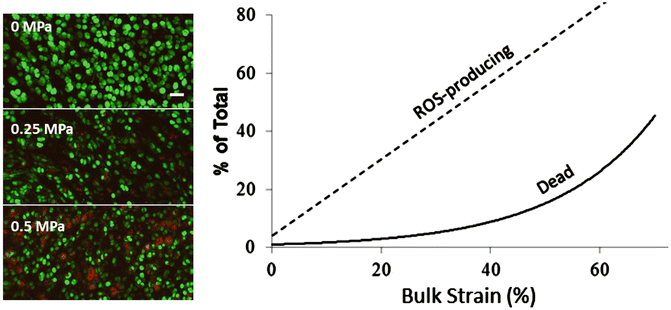
Fig. 17.2
Strain-related ROS and death. Left: representative confocal images show oxidant-producing cells (red) and live cells (green) after compression with the indicated stress magnitudes. The number of red cells increases from 0 to 0.5 MPa. The bar indicates 40 μm. Right: plot summarizing strain effects on ROS production and cell death under bulk tissue strains ranging from 0 to >60 %. The linear correlation coefficient (r 2) for ROS versus strain was 0.87
Explant experiments involving repeated cyclic loading over time frames of 1 week showed potentially pathogenic overloading-related changes in mitochondria. Seven days of repeated high-amplitude loading to strains of 40 % or more reduced mitochondrial potential and respiratory activity as measured by oxygen consumption rate (Fig. 17.3). In contrast, mitochondrial performance was unaffected after 7 days of low-amplitude loads that produced strains of less than 25 %. Gavriilidis and Young recently published results showing that OA chondrocytes exhibit the same constellation of mitochondrial abnormalities including depressed mitochondrial membrane potential and diminished respiratory capacity [42]. The authors also showed substantially lower expression of SOD2 in OA chondrocytes, which may expose mitochondria to oxidative damage. This interpretation was supported by additional findings showing increased proton leakage, which is thought to reflect increased lipid peroxidation in mitochondrial membranes. Interestingly, work in a mouse joint injury model showed that mitophagy, a major mechanism for mitochondrial turnover and repair, is suppressed in overloaded cartilage and treatment with rapamycin, which stimulates mitophagy by blocking the mammalian target of rapamycin, showed chondroprotective effects [43, 44]. These findings suggest that oxidant overproduction from chronic overloading together with deficiencies in SOD activity lead to the mitochondrial derangements present in OA chondrocytes.
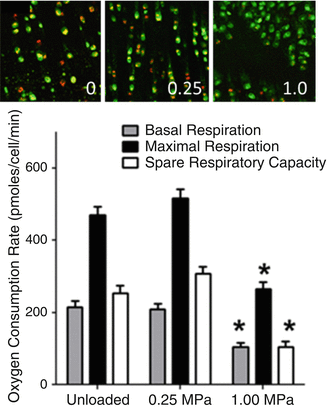
Fig. 17.3
Overloading leads to mitochondrial dysfunction. The top panel shows representative confocal micrographs taken of JC-1-stained chondrocytes after 1 week of loading with the indicated stresses (0, 0.25, 1.0 MPa). The orange and yellow staining indicating mitochondrial polarization was diminished in the 1.0 MPa-loaded explant. The graph in the lower panel shows loading effects on mitochondrial respiration in chondrocytes as measured by oxygen consumption rate. Basal and maximal respiration and spare respiratory capacity were all significantly reduced in the 1.0 MPa group versus the unloaded and 0.25 MPa-loaded groups
The mechanoresponsive oxidant production pathway outlined above seems maladaptive to the extent that it is responsible for oxidative insults to cartilage. However, there is evidence to suggest that the pathway performs a crucial physiologic function by regulating glycolytic ATP synthesis in response to normal loads. Although mitochondria contribute only a small fraction of cellular ATP in chondrocytes, it was found that oxidants formed as a by-product of mitochondrial electron transport are required for glycolytic activity [45]. Cyclic loading of explant cartilage that yielded low- to medium-amplitude strains increased ATP by fourfold over resting controls [39]. Remarkably, this stimulatory effect was abolished by the same antioxidants and transduction inhibitors that suppressed trauma- and overload-induced damage and death. Thus, it appears that the very process that wreaks havoc in overloaded cartilage maintains redox balance and modulates energy production in response to normal loading (Fig. 17.4). These findings also suggest that impairment of ATP production is one of the main consequences of mitochondrial dysfunction of the kind seen in overloaded and osteoarthritic cartilage.
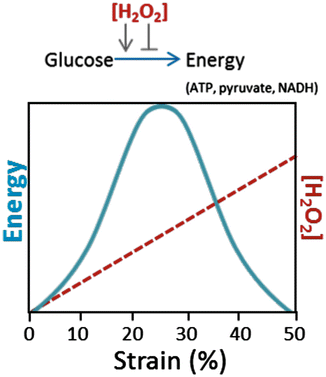
Fig. 17.4
Effect of strain-induced peroxide on energy production. The diagram illustrates the concept that H2O2 resulting from superoxide dismutation has concentration-dependent effects in regulating energy production via glycolysis. Energy production in the form of ATP, pyruvate, and NADH is stimulated at low concentrations produced under moderate strains (10–25 %). As strain increases to 30 % or more, H2O2 formation increases to levels that inhibit glycolysis, resulting in progressive depletion of energy stores
Prospects for New Therapies
The fact that oxidative damage to chondrocytes occurs through a defined mechanotransduction pathway suggests there are diverse opportunities for intervention that include, but are not limited to, antioxidants. The central role of mitochondria in mechanotransduction makes them an obvious candidate for drug development (Fig. 17.5). Indeed, amobarbital, a drug that suppresses O2 •− production by blocking electron transport, has shown some promise in a rabbit cartilage injury model [46], where intra-articular injection improved chondrocyte viability and ATP content (Fig. 17.6). Treating with nocodazole or cytochalasin B, which reduces O2 •− by blocking mechanotransduction at the level of the cytoskeleton, also improved viability and ATP content. Oxidant scavengers also remain on the list of potential therapeutics to moderate overloading effects. N-acetylcysteine (NAC), for example, spared chondrocytes from oxidative death in the rabbit and in a variety of other overloading circumstances. While the low cost and safety of NAC and other common oxidant scavengers add to their appeal as therapeutic agents, antioxidant activity is lost after only one reaction. In contrast, antioxidant enzymes such as superoxide dismutase (SOD) are not consumed by oxidant catalysis and may have more lasting effects. Recent increases in the availability of small and stable SOD mimetics offer promising options as oxidant scavengers [47]. Combinations of drugs such as SOD mimetics and NAC may be used to ensure that the rapid dismutation of O2 •− does not overwhelm the cells’ intrinsic ability to detoxify H2O2 and other secondary oxidants.
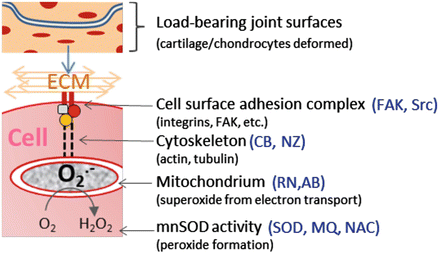
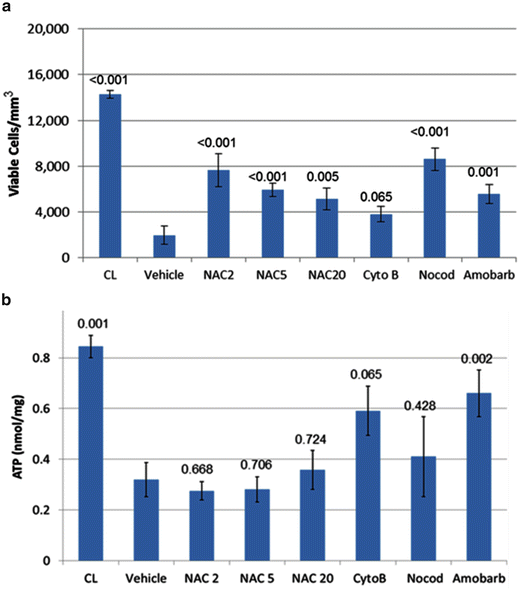
Fig. 17.5
Intervening in chondrocyte mechanotransduction. The diagram illustrates known components of the mechanotransduction pathway and inhibitors that have been shown to block the pathway at various steps. These include FAK and Src inhibitors targeting ECM adhesion, cytochalasin B (CB) and nocodazole (NZ) that block cytoskeleton formation, rotenone and (RN) and amobarbital (AB) that block mitochondrial electron transport, and superoxide dismutase mimetic (SOD), mitoquinone (MQ), and N-acetylcysteine (NAC), which participate in superoxide and peroxide detoxification
Fig. 17.6
Effects of treatment on chondrocyte viability and ATP content in injured cartilage. Rabbits with chondral damage caused by blunt impact were treated after injury. One-week post-op injured cartilage was imaged by confocal microscopy to determine viable cell density (a) and then assayed for ATP content (b). Cartilage from contralateral limbs (CL) was used as uninjured controls. Injured joints were untreated (no Rx) or treated with NAC at 2, 5, or 20 mM (NAC2, NAC5, NAC 20), cytochalasin B (CytoB), nocodazole (Nocod), or amobarbital (Amobarb). Columns and error bars indicate means and standard errors. The numbers above the columns show p-values for treated versus vehicle (one-way ANOVA)
That damaging oxidants can be produced intracellularly by chondrocytes buried in the dense, avascular cartilage matrix presents substantial obstacles to the bioavailability of compounds administered systemically or even intra-articularly. Indeed, negative clinical trial results for some antioxidant therapies may be attributable to poor drug availability in cartilage. Agents that diffuse through the cartilage ECM must also either pass through cellular membranes or be actively taken up by cells to finally reach their targeted site of action. Importantly, the antioxidants and mechanotransduction inhibitors mentioned above penetrate the cartilage ECM and pass readily through cell membranes.
As a note of caution, recent gains in our understanding of chondrocyte mechanobiology make it apparent that over-suppressing ROS can disrupt normal responses to loading that are critical for cartilage homeostasis. Thus, the high levels of antioxidants used to combat oxidative damage in other tissues may not be tolerated in cartilage. This potential for harm underscores the need to carefully optimize dosages and treatment duration in animal models before contemplating testing in people.
References
1.
Sutipornpalangkul W, Morales NP, Harnroongroj T. Free radicals in primary knee osteoarthritis. J Med Assoc Thai. 2009;92 Suppl 6:S268–74.
2.
Ziskoven C, et al. Oxidative stress in secondary osteoarthritis: from cartilage destruction to clinical presentation? Orthop Rev (Pavia). 2010;2(2):23.CrossRef
Related posts:
 Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
 Outcomes of ACL Injury: The MOON Consortium
Outcomes of ACL Injury: The MOON Consortium
 Aging and Post-Traumatic Arthritis
Aging and Post-Traumatic Arthritis
 Biomarkers of PTA
Biomarkers of PTA
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


