Fig. 15.1
Overview of cell death-related pathways. Extrinsic apoptosis involving death receptors and extracellular ligands leads to the activation of caspases. Inhibition of caspases is mediated by various molecules including cellular inhibitor of apoptosis proteins (cIAPs), BCl-2, and Bcl-xL. Intrinsic apoptosis is mediated by a largely mitochondria-centric manner that involves either caspase- or non-caspase-directed cell death. Mitochondrial outer membrane permeabilization (MOMP) leads to mitochondrial transmembrane potential dissipation and to subsequent release of reactive oxygen species (ROS) and an assortment of proteins such as cytochrome c (CYTC), which interacts with other cytoplasmic proteins to form the apoptosome, a multiprotein complex that triggers the proteolytic cascade including caspase 9 and the executioner caspase 3. MOMP also leads to the release of direct IAP-binding protein with low pI (DIABLO), which inhibits cIAPs activity. Other molecules such as apoptosis-inducing factor (AIF) and endonuclease G (ENDOG) act independently from caspase activity and translocate to the nucleus to induce large-scale DNA fragmentation. The release of ROS can lead to necrotic cell death or can trigger survival pathways including autophagy [149] (see Fig. 15.2). Autophagy can also be activated by extrinsic TNF signaling via NF-κβ activation. The location of p53 can initiate different pathways. Following cellular stress or damage, nuclear p53 can directly induce autophagy, while cytoplasmic p53 induces apoptosis (Figure adapted from [18, 150]
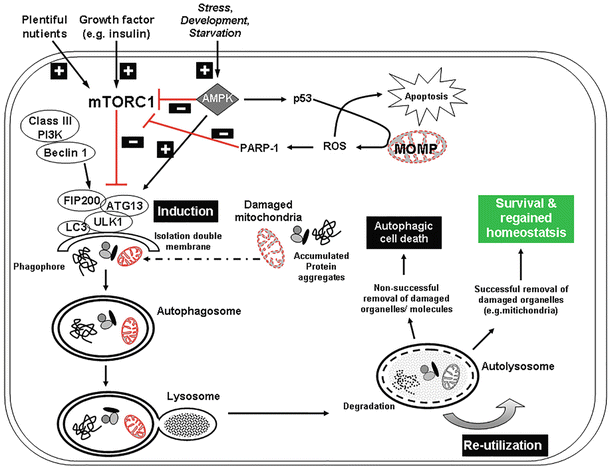
Fig. 15.2
Autophagy pathway in homeostasis and response to stress. As a major degradation pathway in eukaryotic cells, autophagy is essential for the removal of damaged organelles and protein aggregates from the cytoplasm that can lead to cellular dysfunction [151, 152]. Three central kinases are pivotal for controlling cell homeostasis including AMP kinase (AMPK), the mammalian target of rapamycin complex 1 (mTORC1), and Unc-51-like kinase 1 (ULK1). mTORC1 inhibits autophagy by preventing the induction of the double-membrane phagophore and subsequent autophagosome formation by phosphorylating autophagy-related protein 13 (Atg 13) and ULK1, thus preventing the formation of a kinase complex [153–155]. mTORC1 can simultaneously inhibit autophagy and stimulate protein synthesis and cell growth, which can lead to an accumulation of damaged proteins and organelles and hence contribute to damage at the cellular level [156]. Following external or internal stressors, such as injury, starvation, energy depletion, and during development, AMPK can initiate apoptosis via p53 or can inhibit mTORC1’s capacity to inhibit autophagy and activate ULK1, thus initiating higher autophagic activity [27, 157]. As well, ROS can induce PARP-1 activity to reduce mTOR signaling and activate autophagy [149]. The cytoprotective response that autophagy provides is a means for the cell to recover following stress and restore homeostasis. However, when the autophagic response is not sufficient, imbalanced, or excessive, this may lead to cell death [47, 158, 159]
Extrinsic Apoptosis
Cell death is mediated by extracellular stress signals that are detected and propagated by specific transmembrane death receptors (Fig. 15.1). Examples of extrinsic binding of lethal ligands to death receptors include (1) tumor necrosis factor α (TNF-α) by TNF-α receptor 1 (TNFR1), (2) FAS ligand by CD95 ligand (FASL/CD95L), and TNF (ligand) superfamily, member 10 (TNFSF10), or TNF-related apoptosis-inducing ligand (TRAIL) by TRAIL receptors (TRAILR) 1–2 [19–21]. In contrast, the so-called dependence receptors can signal an extrinsic proapoptotic signal, including netrin receptors, which can exert lethal functions when specific ligand concentrations fall below a critical concentration [21].
Ligand and death receptor interaction activates three major lethal signaling cascades leading to caspase activation including (1) activation of the caspase-8 (or caspase-10)-caspase-3 cascade; (2) activation of the caspase-8-tBID-mitochondrial outer membrane permeabilization (MOMP)-caspase-9-caspase-3 pathway; or (3) ligand deprivation-induced dependence receptor signaling followed by (direct or MOMP-dependent) activation of the caspase-9-caspase-3 cascade [18] (Fig. 15.1). Due to a common dependence on caspase activity, these death pathway cascades can be suppressed by the overexpression of viral inhibitors of caspases like cytokine response modifier A (CrmA) [22] or by pan-caspase chemical inhibitors such as N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk) [11, 23].
Intrinsic Apoptosis
Intrinsic induction of apoptosis involves bioenergetic and metabolic catastrophe together with multiple active executioner mechanisms that may or may not include caspase activity (Fig. 15.1). Overall, intrinsic apoptosis is mediated by mitochondrial outer membrane permeabilization (MOMP) and is always accompanied by dissipation of mitochondrial transmembrane potential, a release of mitochondrial intermembrane space (IMS) proteins into the cytosol, and inhibition of the respiratory chain. Caspase activity involvement in intrinsic apoptosis involves MOMP, cytosolic cytochrome c interaction with APAF1 and dATP to form the apoptosome, and triggering of the caspase-9-caspase-3 proteolytic cascade [24]. Independent of caspases, apoptosis-inducing factor (AIF) and endonuclease G (ENDOG) relocate from the mitochondria to the nucleus to mediate DNA fragmentation [25–27] (Fig. 15.1).
Many intracellular stress conditions can trigger intrinsic apoptotic demise, but are principally controlled by or coordinated with the mitochondria. Documented stressors include oxidative stress, DNA damage, cytosolic Ca2+ overload, mild excitotoxicity, and accumulation of unfolded proteins in the endoplasmic reticulum (ER), among others [28].
Regulated Necrosis
While cell death in the form of necrosis can result from a traumatic event, as defined by the absence of apoptotic or autophagic features [17, 29], it is now recognized that necrosis can also occur in a regulated manner [30–34] in many physiological and pathological contexts [35]. Under specific circumstances, the induction of regulated necrosis or “necroptosis” can be induced by several ways including ligation of death receptors (e.g., TNFR1), excitotoxicity, and alkylating DNA damage, involving receptor-interacting protein kinase (RIP1) RIP3 and caspase-8 [30–34, 36, 37].
Autophagic Cell Death
Autophagy is a controlled cellular catabolic process that is critical for cell survival, differentiation, development, and homeostasis that involves the formation of double-membrane vesicles, called autophagosomes [38, 39] (Fig. 15.2). Autophagosomes deliver cytoplasmic contents (typically unwanted organelles or protein aggregates) to lysosomal machinery for degradation and recycling. In addition to homeostasis, the autophagic pathway also has a cytoprotective role as it is stimulated to counter various cellular or subcellular stresses for cell survival including reactive oxygen species (ROS), DNA damage, dysfunctional organelles, hypoxia, protein aggregates, or intracellular pathogens [40] (Fig. 15.2). In skeletal tissues, the autophagic system enables osteoblasts, osteoclasts, and chondrocytes to survive within the local hypoxic, and even hypertonic environment, and to overcome the presence of stressors and nutrient deficiencies. However, under situations of chronic stress or certain physiologic and pathologic states, cell death by autophagy can occur [17, 39, 41–44] and in most cases due to an inhibition or reduced activity of this process [45]. Dysregulation of this process has been associated with a variety of human pathophysiological processes, such as aging, cancer, cardiovascular diseases, neurodegenerative disorders, and OA [46, 47].
Mitotic Catastrophe
Although mitotic catastrophe has been thought to be a separate mode of cell death, more recently it has become evident that mitotic catastrophe does not execute cell death in the pure sense, but rather it represents an oncosuppressive mechanism that leads to either apoptosis, necrosis, or cell senescence to eliminate mitosis-deficient and genomically unstable cells [48].
Anoikis or Oncosis
Another distinct form of cell death is principally regulated by changes or loss in adhesion between integrins and the epidermal growth factor receptor (EGFR) and the extracellular matrix [49, 50]. Anoikis displays some features associated with necrosis, including increased membrane permeability, cell and organelle swelling, and absence of internucleosomal DNA fragmentation [51, 52]. However, the inhibition of extracellular signal-regulated kinase 1 (ERK1) signaling and an overexpression of the BCL-2 family member BIM [50, 53] can be used to identify this phenomenon. It is important to note that downstream of anoikis-initiated cell death, the same molecular machinery for intrinsic apoptosis is utilized (Fig. 15.1). Cell death resembling anoikis has been observed in ischemic heart disease [54], in atherosclerotic lesions [55], and in both cartilage and bone [56].
Chondroptosis
In cartilage tissue, Roach et al. used the term “chondroptosis” to indicate a specific form of chondrocyte apoptosis [57] that involves altered protein synthesis as observed by an increased number of endoplasmic reticulum (ER) and Golgi apparatus, which appears distinct from typical receptor-mediated or mitochondrial pathways. The ER membranes appear to segment the cytoplasm to produce autophagic vacuoles in the cytoplasm where organelles are digested and disposed into the lacunae. This divergent cell death process is consistent with the avascular nature of cartilage, where chondrocytes are isolated within their lacunae and cannot rely on the phagocytic removal or apoptotic cell remnants typical of other tissues [58].
Mechanisms of Post-Traumatic Cell Death
Mechanical Stress
Mechanical injury is known to trigger cell death, and the resultant cartilage matrix degradation has been reported in animal [11, 59–63] and human cartilage [11, 64–66]. Acute impact or more chronic repetitive injury can significantly reduce cell viability. In human cartilage explants, a single episode of mechanical injury increased the number of apoptotic cells in a time-dependent pattern, which was inhibited by incubating the impacted explants with the pan-caspase inhibitor z-VAD-fmk [11]. Repetitive trauma can also induce apoptosis in vitro [67]. In vivo, the cartilage degeneration induced by repetitive injury (anterior cruciate ligament transection in rabbit knees) was significantly reduced with caspase inhibition supporting a potential therapeutic role [23]. Other studies also demonstrated the potential therapeutic role of caspase inhibitors [11, 66], BMP7 [68], FGF-18 [59], P188 surfactant [65], and inhibitors of focal adhesion kinase (FAK) and Src family kinase (SFK) [69] for chondroprotection after impact trauma or tissue injury as a result of a surgical procedure [70].
Early cell death was reduced by a nonionic surfactant (P188), which suggested the loss of cell membrane integrity as one mechanism by which blunt trauma can affect cell viability [71]. P188 has also been shown to affect stress-related p38 signaling and the inhibition of GSK3 and IL-6 suggesting other pathways leading to cell death [72].
Autophagy
Autophagy involves recycling of long-lived protein organelles and is a critical process for cellular homeostasis and cytoprotection. Cell stress stimulates autophagy, for example, autophagy is upregulated in response to ischemia/reperfusion and pressure overload in the heart [73, 74]. Therefore, autophagy may be similarly affected in mechanoresponsive tissues such as cartilage. Recent observations indicate that there is a basal level of autophagy in normal cartilage and this is increased in response to nutrient deprivation [75]. In aged and OA-affected human articular cartilage and in animal models of OA, such as surgically induced joint instability, there is a marked reduction in the expression of important proteins that regulate the autophagy pathway including ULK1, Beclin1, and LC3, which was accompanied by increased apoptosis [76] (Fig. 15.2) and may also play a role in programmed cell death implying complex interactions.
Several instances of crosstalk are known between members of autophagy and apoptosis pathways [77, 78]. The autophagy protein, Atg5, induces mitochondria-based apoptosis, while Bcl-2 overexpression protects against Atg5-mediated mitochondrial dysfunction. Beclin 1, an essential autophagy protein, is regulated by the Bcl-2 proteins and in normal conditions. Bcl-2 and Bcl-XL suppress autophagy by associating with Beclin 1 [79]. Reduced Beclin 1 heterozygous mice (Beclin 1+/–) have reduced autophagy and apoptosis and heart infarct size after I/R injury [80], suggesting that Beclin 1 might be actively involved in mediating apoptosis.
LC3-II and Beclin-1 are both expressed by chondrocytes in the maturing region of the growth plate. In the hypertrophic zone, there is reorganization of these proteins into punctate granules that are characteristic of the autophagolysosome, while TEM studies indicated the presence of double-membrane autophagosomes. Once mineralization has begun, autophagic changes in hypertrophic chondrocytes are seen followed by the initiation of programmed cell death [81]. Further research into the relationship between mechanical injury, apoptosis, and autophagy may reveal additional targets to preserve articular cartilage viability.
Oxidative Damage
Mechanical injury can release reactive oxygen species (ROS). ROS are formed during metabolism of oxygen and play important roles in cell signaling. ROS activity is dynamically balanced by enzymatic and nonenzymatic antioxidants, which act by inhibiting oxidative enzymes or via scavenging free radicals [82]. Increased levels of ROS (oxidative stress) can be damaging and impact cell survival. Mitochondria are a source of post-injury ROS, and chondrocytes express enzymes (such as NADPH oxidase and iNOS) that generate superoxides. Superoxide release can occur within five minutes of impact injury [83]. Decreased mitochondrial superoxide dismutase within OA chondrocytes affects chondrocyte intracellular metabolism [84], and superoxide dismutase mimetic increased cell viability after injury [85]. Further indirect evidence of the oxidative cell death is provided by reports of antioxidant treatment that enhanced cell viability after injury [83, 85]. The antioxidant resveratrol also protected against IL-1beta-induced catabolic effects and prevented IL-1 induced chondrocyte apoptosis via its inhibition of mitochondrial membrane depolarization and ATP depletion [86]. Finally, oxidative stress can have implications beyond post-traumatic cell death. Antioxidative capacity diminishes in degenerating regions of OA cartilage, and the resulting oxidative stress induces replicative senescence and telomere genomic instability [87]. Inadequate control of ROS may be one of the central factors in OA pathophysiology.
Extracellular Matrix
Chondrocytes secrete and maintain a spectrum of proteins serving multiple functions in addition to structural support, such as providing a reservoir for cytokines and growth factors that regulate cell behavior, proliferation, and differentiation, all providing cues that are critical for cell survival [88–90]. Loss of glycosaminoglycan can indirectly predispose cells to necrosis or programmed cell death following mechanical injury [91]. Significant changes in the structure and composition of the perichondrocytic environment during the progress of PTOA can alter the transmission of physical forces to the cell as well as biochemical stimuli that regulate cell response [92]. Ongoing degeneration, deterioration, disintegration, and erosion of matrix lead to an expanding zone of cell death that further intensifies tissue degeneration. Profound changes in adhesion molecules and ECM signaling via receptors can directly activate apoptotic pathways (e.g., FAS and TNF-αR), induce anoikis through loss of cell adhesion, or secondarily induce apoptosis, for example, via cytoskeletal changes [93–96].
Anoikis is a variant of apoptotic cell death as a result of lost, reduced, or inappropriate cell adhesion (see review by [92]). Despite the initiation via changes in the ECM, the downstream apoptotic process is mediated through pathways that converge to activate caspases. Several ECM proteins are essential for cell survival; among them collagen type II is the most critical in maintaining chondrocyte viability and preventing apoptosis [97]. Integrin binding to ECM components is also important for cell survival. Proteins important for binding to integrin receptors are laminin, fibronectin, and collagen types II and IV [88, 98]. Several studies directly implicate integrin-ligand interactions in chondrocyte death. For example, blocking antibodies against the integrin α5 subunit (CD49e) induced death in human chondrocytes [99]; RGD peptides reduced cell viability in cultured chicken chondrocytes [100] and induced apoptosis in cultured chondrocytes and in cartilage explants [101]; and type X collagen deposition and chondrocyte survival in chicken sterna are dependent on CD49b and CD49c integrin subunits [93].
Although ECM proteins are important for cell survival, it is well known that ECM protein fragments have deleterious effects: the more common response being inflammatory and catabolic. For example, the 29 kDa fragment of fibronectin induces inflammatory responses [102] and catabolic proteases such as matrix metalloproteinase-13 (MMP-13) [103]; fragments of hyaluronan generate nitric oxide [104] and can increase MMP-13 production [105]. However, a synthetic peptide fragment of type II collagen (residues 195–218) (CB12-II) without the RGD sequence induced apoptosis in bovine cartilage explants in a manner related to chondrocyte hypertrophy [106, 107].
Mitochondrial Damage
The evidence for mitochondrial involvement in cell death is overwhelming (see reviews [108–111]). Mitochondrial dysfunction has been linked to apoptosis, aging, and OA [82, 112–115]. Although IL-1 is not generally considered a proapoptotic signal, IL-1 can induce apoptosis in chondrocytes by inducing mitochondrial dysfunction and depleting cellular energy stores [114]. In addition to the mitochondrial involvement in NO-induced apoptosis [116], oxidative stress and mitochondrial dysregulation play an important role in the initiation and progression of OA [117, 118]. Mitochondria are involved in epiphyseal chondrocyte death during bone development and are associated with maturation-dependent reduction of oxidative phosphorylation [119] and changes in Bcl-2 protein levels [120]. The causal relationship between phosphate ions, NO production, and mitochondrial dysfunction in growth plate chondrocytes suggests a similar mechanism in PTOA [121, 122].
p53 and c-Myc
p53 is commonly involved in increased cell death in aging cartilage. In arthritic cartilage, DNA fragmentation positively correlated with p53 [123]. p53 increased in articular cartilage with aging and was associated with decreased viable cell density in rabbits [124]. p53 is also implicated in nitric oxide-induced cell death via p38 MAP kinase and NF-κβ supporting an active role in chondrocyte survival [125]. In cultured human chondrocytes, hydrostatic pressure-induced apoptosis was associated with increased p53 expression [126].
c-Myc is associated with hypertrophic differentiation of chondrocytes [127, 128] and frequently colocalized with cells containing breaks in DNA strands. c-Myc levels increased in areas of cartilage erosions in canine models of OA [129] and colocalized with apoptotic cells in human arthritic cartilage [123]. c-Myc has also been implicated in apoptosis induced by hydrostatic loading [126]. These findings suggest that, in addition to developmental cell death, p53 and c-Myc may also regulate injury- and OA-related chondrocyte death.
Therapeutic Targets
Despite the common etiology of joint trauma, the multifaceted nature of development and progression of post-traumatic arthritis indicates that effective therapy will likely involve multiple approaches. Matrix homeostasis relies on a balance between net anabolic and catabolic activities, which are directly influenced by the number of available chondrocytes. The weight of existing evidence offers chondrocyte death as an excellent target for therapeutic intervention in OA. To achieve prophylactic and therapeutic success, further research into chondrocyte death, cartilage degeneration, and arthritic progression is required. Several potential treatments are being actively translated to clinical application.
Inhibition of Apoptosis
Caspase inhibitors and other chondroprotective agents such as BMP7 [68] are being translated for clinical use. Caspase inhibition is being pursued in several diseases, including acute and chronic neurodegenerative diseases, myocardial infarction, and liver apoptosis [130–132]. Application of these clinically relevant inhibitors may facilitate translation into the treatment of acute post-traumatic joint injuries. A proof of principle for the efficacy of caspase inhibitors in attenuating cartilage damage has been provided in vitro and in animal models [11, 23, 66]. Mechanical injury to chondrocytes and cartilage explants is associated with caspase activation, and caspase inhibitors reduce ECM damage and protect against cell death. Studies with cartilage explants showed that the cartilage superficial zone is where the highest level of cell death occurs. Reducing damage in this zone that is critical for cartilage integrity by the use of caspase inhibitors would be expected to reduce the severity of PTOA, and this has been demonstrated in animal models. A more precise identification of the intrinsic and extrinsic mechanisms leading to chondrocyte death is also ongoing to broaden the spectrum of antiapoptotic targets for therapeutic interventions.
Antioxidant Treatment
ROS can lead to accelerated ECM degradation and cell death. Therefore, the use of antioxidants or enhancement of superoxide dismutase to combat excess ROS has promise [133, 134]. A superoxide dismutase mimetic increased cell viability after injury in cartilage explants [85], and N-acetylcysteine reduced postimpact chondrocyte death in osteochondral explants [12]. Rotenone, an electron transport chain inhibitor, reduced superoxide released by mitochondria after injury and enhanced cell survival [83]. Resveratrol is a naturally occurring polystilbene (to which the health benefits of red wine have been attributed). Resveratrol has an antiapoptotic effect in osteoarthritic chondrocytes stimulated with IL-1, mediated presumably via inhibition of the induction of PGE2 [86].
Autophagy Targets
Reduced expression of autophagy proteins has not only been observed in the knee cartilage of mice with joint instability but also in cartilage explants that were subjected to single-impact mechanical injury [76]. In such models, a time-dependent increase in cell death and ECM damage has been documented. The autophagy pathway is controlled through diverse mechanisms and signaling pathways, and a large number of pharmacological activators and inhibitors of autophagy have been identified [135]. The kinase mammalian target of rapamycin (mTOR) is a key regulator of autophagy and integrates signals from various extracellular stimuli. Cartilage-specific ablation of mTOR enhanced autophagy and protected against OA development in mice [136]. Rapamycin, an inhibitor of mTOR and an activator of autophagy, reduced the severity of degenerative changes in a mouse model that is associated with increased chronic mechanical load [137]. In the cartilage explant model, rapamycin also protected against injury-induced cell death and ECM damage. These observations suggest that deficits in autophagy contribute to cell death and ECM damage after joint injury. The availability of autophagy activators in clinical use, such as rapamycin, renders autophagy as a promising therapeutic target in PTOA.
Cell Membrane-Stabilizing Surfactants
Poloxamer surfactants are water-soluble copolymers with hydrophobic and hydrophilic chains that insert directly into and repair damaged cell membranes [71]. Poloxamer 188 (P188) increased the percentage of cell viability in vitro and in vivo within four days of injury, increasing cell density in vivo after 6 weeks [71, 138, 139]. In addition to increasing cell viability in injured cartilage, P188 also prevented secondary cell death in the uninjured tissue adjacent to the site of injury [65].
Cartilage Progenitor Cells
Despite the lack of vascularity, articular cartilage contains distinct populations of progenitor cells [140–148], most of which are found in the superficial zone [141, 142, 148]. Mechanical injury commonly induces cell death that is predominantly concentrated in the superficial zone. Losing the majority of progenitors as a consequence of injury may impact tissue homeostasis, since these cells are likely to be more suited to repair injured or degenerated tissue than the terminally differentiated chondrocyte. Preserving the progenitor subpopulation or utilizing these cells in cell therapy or tissue engineering could expand our therapeutic toolset for preventing post-traumatic OA.
Summary/Conclusion
The occurrence of cartilage cell death following joint injury has been documented in human tissue samples, in animal and in vitro models. There is also a close relationship between cell death and ECM damage, which appear to potentiate each other and thus generating a key mechanism that mediates the chronic process that ultimately manifests as PTOA. Although incomplete, there is a growing understanding of causes, mechanisms, and types of cell death following injury, and this has led to the identification of several promising therapeutic targets which have been validated in vitro and in animal models. The link between chondrocyte death and PTOA remains to be conclusively proven. The development of sustained release formulations of drugs such as caspase inhibitors or autophagy activators for intra-articular application holds great promise to reduce the effects of injury, thereby potentially reducing the risk for PTOA in the patient with joint injury.
References
1.
Horton Jr WE, Bennion P, Yang L. Cellular, molecular, and matrix changes in cartilage during aging and osteoarthritis. J Musculoskelet Neuronal Interact. 2006;6:379–81.PubMed
2.
Stevens AL, Wishnok JS, White FM, Grodzinsky AJ, Tannenbaum SR. Mechanical injury and cytokines cause loss of cartilage integrity and upregulate proteins associated with catabolism, immunity, inflammation, and repair. Mol Cell Proteomics. 2009;8:1475–89.PubMedCentralPubMed
3.
Iannone F, Lapadula G. The pathophysiology of osteoarthritis. Aging Clin Exp Res. 2003;15:364–72.PubMed
4.
Martel-Pelletier J, Boileau C, Pelletier JP, Roughley PJ. Cartilage in normal and osteoarthritis conditions. Best Pract Res Clin Rheumatol. 2008;22:351–84.PubMed
5.
Layton MW, Goldstein SA, Goulet RW, Feldkamp LA, Kubinski DJ, Bole GG. Examination of subchondral bone architecture in experimental osteoarthritis by microscopic computed axial tomography. Arthritis Rheum. 1988;31:1400–5.PubMed
6.
Revell PA, Mayston V, Lalor P, Mapp P. The synovial membrane in osteoarthritis: a histological study including the characterisation of the cellular infiltrate present in inflammatory osteoarthritis using monoclonal antibodies. Ann Rheum Dis. 1988;47:300–7.PubMedCentralPubMed
7.
Kaneko S, Satoh T, Chiba J, Ju C, Inoue K, Kagawa J. Interleukin-6 and interleukin-8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines Cell Mol Ther. 2000;6:71–9.PubMed
8.
Goldring SR. The role of bone in osteoarthritis pathogenesis. Rheum Dis Clin North Am. 2008;34:561–71.PubMed
9.
Radin EL, Rose RM. Role of subchondral bone in the initiation and progression of cartilage damage. Clin Orthop Relat Res. 1986;213:34–40.PubMed
10.
Quinn TM, Allen RG, Schalet BJ, Perumbuli P, Hunziker EB. Matrix and cell injury due to sub-impact loading of adult bovine articular cartilage explants: effects of strain rate and peak stress. J Orthop Res. 2001;19:242–9.PubMed
11.
D’Lima DD, Hashimoto S, Chen PC, Colwell Jr CW, Lotz MK. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthritis Cartilage. 2001;9:712–9.PubMed
Related posts:
 Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
 Closed Joint ACL Disruption Murine Model of PTA
Closed Joint ACL Disruption Murine Model of PTA
 Biomarkers of PTA
Biomarkers of PTA
 Instability: Dynamic Loading Models
Instability: Dynamic Loading Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


