CHAPTER 18
Pharmacologic Management of Spasticity: Oral Medications
Jay M. Meythaler and Riley M. Smith
Spastic hypertonia encompasses a variety of conditions that may contribute to increased tone or involuntary movement, including dystonia, rigidity, myoclonus, muscle spasm, posturing, and/or spasticity (1). The neurologic localization of the lesion that is the cause of spastic hypertonia may result in the different clinical manifestations noted. The other conditions associated with spastic hypertonia, such as the “clasp knife phenomenon,” Babinski or Hoffman reflexes, rigidity, and particularly acquired dystonia, may be more disabling than spasticity in acquired brain injury (2,3), some cases of cerebral palsy (CP) (4), and multiple sclerosis (MS) (5). For this reason, they are often considered to be part of the broader “upper motor syndrome,” of which spasticity is a part (6).
One of the hallmarks of upper motor neuron injury or destructive process above the conus medularis is the development of spastic hypertonia. It is one of the most disabling aspects of central nervous system (CNS) injury or illness. Spastic hypertonia can interfere with the functional use of remaining motor function, limit the range of motion of a joint, or cause pain. This can be very disabling with regard to mobility, transfers, activities of daily living, sitting, or sleep (7–10).
However, there are beneficial aspects of spastic hypertonia. Spasticity can aid in maintaining muscle tone and mass, resulting in increased blood return in the venous system, and aiding in reflexive bladder and bowel emptying (11,12). There has been evidence that spasticity decreases the incidence of osteoporosis in paralyzed extremities; however, this has been inconsistent and most studies indicate spasticity is not protective of bone mineral density (13–17). The increased muscle tone can also provide a stabilizing torque at various joints to improve posture, aid in sitting, or aid with transfers. The increased muscle mass over bony prominences can help prevent decubitis ulcers. Hence, spastic hypertonia is not an entity that should be completely eliminated, just modulated to improve pain and function.
In general, oral medication has been less successful in managing spastic hypertonia after acquired cerebral injury than in spinally related causes of spastic hypertonia (8,18). Furthermore, all the potentially useful drugs for spasticity have associated central side effects that need to be carefully weighed when considering their use. The pharmaceutical management of spastic hypertonia after CNS injury or illness has historically been confined to the use of five primary medications: baclofen, diazepam, dantrolene sodium, clonidine, and tizanidine (1,6,8,19–22). Over the past several years, there has been an increasing body of research in cannabis and cannabis-derived medications (23,24). There is limited evidence that other medications may be useful as well.
Spastic hypertonia clinically presents differently when the lesions are in the spine, brainstem, or cerebral cortex (3,10). The precise neurologic localization of the lesion can result in different clinical manifestations of spasticity (10). However, one does not treat spastic hypertonia based on the anatomic location of the lesion. Rather, treatment is focused on the clinical presentation of the patient. Furthermore, it is clear that some medications may be preferred for upper limb spasticity, whereas others may be preferred for lower limb spasticity (5,22). When one considers the cognitive and other side effects of medications, one needs to customize the treatment of medications quite precisely.
Determining the benefit of oral medications for the treatment of spastic hypertonia must focus on evaluating the patient with physiologic and functional outcome measures (25). The most commonly employed scales are the Ashworth Scale, the Modified Ashworth Scale, the Spasm Frequency Scale, and reflex scores (26) (as seen in Tables 18.1 and 18.2). These scales are those that have been most often accepted by the Food and Drug Administration (FDA) for pharmaceutical and investigational trials to obtain a clinical indication for use in “spasticity.” They are also easily administered in a clinical setting. In recent years, there has been increased criticism toward the Ashworth and Modified Ashworth Scales due to limited reliability and validity (27). Many studies have begun to use a patient-reported 0–10 numeric rating scale for spasticity, as it is felt that this is more sensitive to changes in the patient’s ability to perform activities and changes in pain (28). These scales are readily applied without special equipment in any clinic, which makes them clinically useful.
TABLE 18.1
DEFINITION OF ASHWORTH, MODIFIED ASHWORTH, SPASM FREQUENCY, AND REFLEX SCORES | |
Ashworth score | |
1 | No increase in tone |
2 | Slight increase in tone, giving a “catch” when affected part is moved in flexion or extension |
3 | More marked increase in tone, but affected part easily flexed |
4 | Considerable increase in tone; passive movement difficult |
5 | Affected part rigid in flexion or extension |
Modified Ashworth score | |
0 | No increase in tone |
1 | Slight catch and release, or minimal resistance at end range |
1+ | Catch, followed by minimal resistance throughout the remainder (less than half) of the ROM |
2 | More marked increase in tone through most of the ROM |
3 | Considerable increase in tone; passive movement difficult |
4 | Affected part(s) rigid in flexion or extension |
Spasm frequency score | |
0 | No spasms |
1 | Mild spasms induced by stimulation |
2 | Infrequent full spasms occurring less than once per hour |
3 | Spasms occurring more than once per hour |
4 | Spasms occurring more than 10 times per hour |
Reflex score | |
0 | Reflexes absent |
1 | Hyporeflexia |
2 | Normal |
3 | Mild hyperreflexia |
4 | 3 or 4 beats clonus only |
5 | Clonus |
Definitions and rating scales used to evaluate muscle tone (1,26), spasm frequency (2), and deep tendon reflexes (2,3).
ROM, range of motion.
In addition, the physician should carefully monitor other medications, including “over-the-counter” medications due to potential interactions and the effects these medications may have on spastic hypertonia. This resource will present in detail the most frequently utilized oral medications and outline briefly the variations in their recommended uses in cerebral disorders versus spinal disorders and when attempting to treat upper extremity spastic hypertonia versus lower extremity spastic hypertonia.
TABLE 18.2A
TABLE 18.2B


A wide range of potential treatments have been investigated. The clinical pharmaceutical management of spastic hypertonia after CNS injury or illness has generally been confined to the use of centrally acting drugs mediated through gamma-aminobutyric acid (GABA) central alpha activity (9,29,30) and peripherally acting drugs that inhibit the release of calcium from the sarcoplasmic reticulum (dantrolene) (20) as well as those that inhibit excitatory neurotransmitter release. We will organize the discussion of these medications by the neurotransmitter, through which they mediate their effect.
GABA AGONISTS
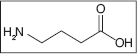
GABA receptor sites are widely present in the CNS, and GABA is the third most prevalent neurotransmitter in the CNS. GABA is present in an estimated 60% to 70% of all synapses in the brain and is inhibitory on a variety of neural pathways associated with spasticity (10,29,31). GABA is always an inhibitory neurotransmitter, affecting both presynaptic and postsynaptic inhibitions. GABA is a completely hydrophilic neurotransmitter and therefore is infinitely lipophobic with a complete inability to cross the blood–brain barrier (32–34).
GABA receptors are distributed in three main receptor subtypes, A, B, and C, with GABA-A and GABA-B most clearly involved in the treatment of spastic hypertonia. GABA-A receptors are a heteropentameric molecule that has at least three distinct subunits (35). The GABA-A receptor is a ligand-gated chloride ion channel that when activated allows increased chloride ion influx, causing membrane hyperpolarization (36,37). GABA-B receptors are G-protein coupled and act on voltage-gated calcium channels, including N-methyl-D-aspartate (NMDA) channels (29,38,39).
G-proteins also increase receptor-operated potassium channel conductance (30). Both mechanisms cause hyperpolarization of the motor horn cells, which is often cited as the predominant mechanism for the antispasticity effects associated with GABA-B agonists (6,29).
The GABA-B receptors definitely are involved in spasticity treatment (3,6,18,32,33), whereas the A receptors have some effects on spasticity modulation and have an additional role in seizure management and anxiolysis (3,6). Because GABA crosses poorly into the CNS across any membrane (33,34), drugs have been developed to either indirectly facilitate the release of GABA or substitute for GABA in the CNS as analogs. It has been established that GABA inhibits the release of dopamine, norepinephrine (NE), acetylcholine, and serotonin (5).
BACLOFEN
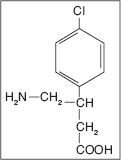
Baclofen is structurally similar to GABA and binds to presynaptic GABA-B receptors within the brainstem, dorsal horn of the spinal cord, and other CNS sites (40–44). Baclofen is a small molecule with a formula weight of 213.67 and the chemical name 4-amino-3(p-chlorophenyl) butyric acid (C10H12ClNO2—see the chemical structure). It is a white to off-white, virtually odorless, crystalline powder with a slightly bitter taste. Structurally, it is very similar to GABA. It is stable as a solid and in solution at room temperatures and decomposes into a lactam at temperatures above 50°C. It was developed in the 1970s and has been a mainstay in the treatment of spastic hypertonia.
Baclofen depresses both monosynaptic and polysynaptic reflexes by blocking the release of neurotransmitters (42,45). This results in inhibition of gamma motor neuron activity to the muscle spindle intrafusal fibers (29). Hence, baclofen inhibits monosynaptic and polysynaptic reflexes (41,46). Because these reflexes can facilitate spastic hypertonia, inhibition of these reflex pathways reduces the overactive reflex response to muscle stretching or cutaneous stimulation. Some cases of dystonia also appear to respond to GABA agonists (2,4,5). Baclofen has some supraspinal activity that may contribute to clinical side effects.
Orally delivered baclofen reaches relatively low concentrations in the spinal cerebrospinal fluid, even after large oral doses (47). Thus, many patients experience central side effects, such as drowsiness, confusion, or attentional disturbances at the dosages required to reduce spasticity (6,40,48,49). Other central effects of the drug include hallucinations, ataxia, lethargy, sedation, and memory impairment (40,48–51). Furthermore, those patients with cerebral lesions are felt to be more prone to the centrally mediated side effects of medication (1). There is a significant reduction in attention span in some traumatic brain injury (TBI) patients who have been placed on oral baclofen, even when testing was done after several months on the medication (52). These effects have been reversible with a withdrawal of baclofen. Because cognitive function is critical for survival and adaptation after CNS injury or illness, any impairment of cognition due to medication may significantly adversely effect the severely brain-injured patient. The sudden withdrawal of baclofen, including baclofen delivered intrathecally, may lead to seizures and hallucinations (12,42).
Pharmacokinetics
Oral baclofen is relatively well absorbed with the peak effect within 2 hours of ingestion (52,53) and a half-life of between 2.5 and 4 hours (6,53), but it has a tissue half-life in the CNS estimated to be 3 to 5 hours (53). The drug is excreted unchanged by the kidney, and only 6% to 15% is metabolized by the liver (29,53). A study in the pediatric population revealed a half-life of 4.5 hours (54).
Treatment
Baclofen is often initiated as a first-line treatment for spasticity and it is endorsed by several guidelines (55,56). The initial dosage of baclofen for spastic hypertonia should be started at 15 mg/d and increased every 3 days by 15 to 20 mg/d at least three times per day due to the relatively short half-life (29,57). For children with CP and at least 2 years of age, it can be started at 2 mg/kg/d divided three times per day until a maximum tolerated dose is reached or 20 mg four times daily (54). Baclofen is often the first drug of choice in spinal causes of spastic hypertonia of spinal origin (6,52). It has been utilized predominately for lower limb spastic hypertonia in spinal cord injury (SCI) (37,58–63) and MS (64–76). Most of these studies on the oral use of the medication have been open-label studies predominately focused on its effects on the lower extremities. The use of baclofen in the upper extremities is based mostly on individual physician experience. There have been suggestions that baclofen may improve maladaptive and descending plasticity, but this is mostly in animal models and intrathecally (77).
In spasticity due to cerebral origin, the use of baclofen is less well established. There are a few small randomized studies on the use of oral baclofen in CP (60,61). In stroke and TBI, there is a single open-label trial (5). This one trial established that baclofen was effective physiologically in improving spastic hypertonia as measured by the Ashworth score in the lower limbs (5). Based on the observed decrease in tone, the authors hypothesize that there was a reduced receptor density for GABA-B receptors in the cervical and brainstem regions than the thoracic region (5). Further investigation may delineate if there is a difference of oral baclofen’s effectiveness on the upper extremities versus the lower extremities.
Seizures are a serious side effect of the use of baclofen and appear to lower the seizure threshold, regardless if the dose of baclofen is increased or decreased (78–82). Other complications of oral baclofen include euphoria, hallucinations, ataxia, memory impairment, lassitude, urinary problems, muscle weakness, dizziness, hypotension, sedation, and blurred vision (3,29). Motor weakness is usually the result of an overly aggressive dosing regimen on the medication or when patients were utilizing their reflexive spastic hypertonia for functional status. The most serious side effects involve abrupt withdrawal, which is characterized by increased spastic hypertonia, increased deep tendon reflexes, pruritic symptoms, autonomic instability, hallucinations, central fevers, seizures, and death (29). These side effects are most likely related to a serotonin syndrome and usually develop within 2 and 6 days after abrupt withdrawal regardless of the delivery method of baclofen (12,42,79–83).
BENZODIAZEPINE DERIVATIVES
Diazepam
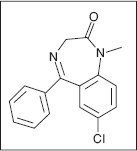
Clonazepam
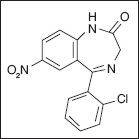
Benzodiazepines were first developed in the 1930s, although clinical usage did not begin until the 1960s (84,85). Diazepam is the prototypical benzodiazepine of this class of compounds characteristic of the 1,4-nitrogenous ring structures most commonly utilized in the United States (84). Diazepam and clonazepam are both documented to be an effective treatment for the initial treatment of spastic hypertonia, but all benzodiazepines (eg, ketazolam, tetraepam) have been useful in the treatment of spastic hypertonia (29,84). They differ primarily with regard to the half-life and the cognitive side effects because some are more sedative, whereas others have a more profound hypnotic effect, and this has been the primary differentiation of these medications.
Benzodiazepines do not directly bind to GABA receptors, but instead promote the release of GABA from GABA-A neurons by facilitating sodium conductance (29,86–89). This results in enhanced presynaptic inhibition and is likely why they are useful in epilepsy (84).
These drugs are all CNS depressant medications and are noted for their antianxiety, hypnotic, antispasticity, and antiepileptic properties. The CNS effects are attributed to their ability to enhance GABA-mediated presynaptic inhibition in the CNS and to depress neuronal activity in the descending lateral and ascending reticular system. This also results in their prominent side effects of sedation and lethargy (84,86,87). These drugs also impair coordination and prolonged use can lead to physical and psychological dependence and/or abuse. Their use should be limited in the elderly and the stroke population (88). Effective dosages vary considerably, and upper level doses are primarily limited by tolerance to side effects. Because clonazepam and diazepam are the prototypical medications, they are the focus of this review.
Pharmacokinetics
Diazepam is quickly absorbed, reaches peak serum levels within 1 hour, and has a half-life approaching 60 hours (90,91). A slow-release preparation does not reach peak serum levels until 3.6 hours after oral delivery (92). Clonazepam reaches peak serum levels in 1 to 4 hours (93,94) and a 30- to 40-hour serum half-life in adults (95), but only 22- to 33-hour serum half-life in children (96). Clonazepam is primarily metabolized in the liver to glucoronides (94), as is diazepam (84,90,91).
Treatment
Diazepam is the prototypical medication utilized in adult spastic hypertonia, and clonazepam, by tradition, is utilized more frequently in pediatric patients. However, there are many who use clonazepam in adults and vice versa. There are many clinicians who employ other benzodiazepines.
Diazepam is well established in the treatment of spastic hypertonia in MS with several clinical trials in the literature (59,70–72,97–99). Diazepam has been traditionally utilized for years in SCI even though the data are less well established, with only a few small studies (59,100) also in mixed populations of patients with SCI, MS, and stroke. Benzodiazepines have more studies establishing their efficacy in cerebral disorders. In stroke patients, diazepam has been reported to be useful (99–103), but the studies have only small numbers of patients and are likely underpowered. In CP, there are several studies supporting the use of diazepam for spasticity especially when associated with pain (56,104). It is the only medication that is listed as probably effective as other medications are very poorly studied (105). There are only a couple of studies documenting the effectiveness of clonazepam in spastic hypertonia: one in a predominately MS population (106) and another small study in pediatric CP patients (107). Considering its widespread use, it is rather quite remarkable that there are so few data. There is relatively more data on the use of ketazolam, which is not available in the United States. However, in these head-to-head studies with diazepam, there was relatively no advantage except less frequent dosing (99,108).
Benzodiazepines are sedative hypnotics and as such tend to exacerbate the complications of memory and alertness in patients with cerebral disorders. Hence, lately, they have not been recommended for use at least in the early stages of recovery. Benzodiazepines such as diazepam and lorazepam also have numerous adverse effects, including generalized CNS sedation with fatigue, drowsiness, and dizziness; paradoxical agitation; confusion; and disorientation with periods of blackouts or amnesia (29,84,86,103). Rapid withdrawal has been associated with irritability, tremors, nausea, and seizures (29,84).
OTHER GABA DERIVATIVES
Gabapentin
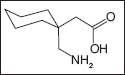
Gabapentin is an analog of GABA. Its chemical name is 1-(aminomethyl)cyclohexaneacetic acid. The mechanism of action of gabapentin continues to be a subject of debate. It is known for certain that gabapentin is not converted metabolically into GABA or a GABA agonist, and it is not an inhibitor of GABA uptake or degradation (109). It most likely does not bind to the GABA receptors (110), but binds at intracellular sites (111). There is some indication that it interacts with amino acid carrier proteins.
Regardless of the mechanism of action, gabapentin does increase GABA turnover, and no general statements can be made on the effect of gabapentin on NMDA receptors. The mechanism by which gabapentin exerts its anticonvulsant effect is not known. However, it appears now to interact with a unique receptor effecting voltage-gated N-type calcium ion channels (112) or possibly to prevent formation of calcium channels in afferent neurons (113). It is generally utilized as an adjunctive medication for the treatment of partial seizures (114).
Pharmacokinetics
Gabapentin is predominately renally cleared, leading to its linear pharmacokinetics (112,115,116). The bioavailability of gabapentin is not dose proportional (117). As the dose is increased, the bioavailability is decreased. Gabapentin is eliminated unchanged from the systemic circulation by renal excretion, and maximum plasma concentration occurs within 2 to 3 hours (118,119) of dosing. Unlike other anticonvulsants, gabapentin apparently does not interfere with the elimination of phenytoin, valproic acid, carbamazepine, and phenobarbital (115).
Treatment
Gabapentin was initially designed as a pharmaceutical agent to treat spasticity, but was found to be a more potent anticonvulsant for which it is approved for marketing in the United States (118,120). Nevertheless, there have been small reports of its effectiveness for spasticity of spinal origin (121–124). However, close analysis of these studies indicates that its effectiveness has been marginal, particularly in terms of a reduction in the Ashworth score. However, some physicians continue to note that some patients with spastic hypertonia have a clinically meaningful response to the medication.
The most frequently noted side effects of gabapentin are somnolence (25%), dizziness (23%), ataxia (20%), nystagmus (15%), headache (14%), tremor (13%), fatigue (12%), and nausea/vomiting (9%), although the frequency is claimed to be less than that noted with most other anticonvulsants (29,115,118). The usage in children has had mixed results, so it is generally only indicated for those more than 12 years of age (114,125). When dosing younger children, there are some pediatric epilepsy dosing guidelines that may be useful that indicate dosing for those 2 years of age or younger using gabapentin syrup 10 mg/kg (126). Dosing for subjects more than 2 years of age received oral capsules based on weight: 200 mg for 16 to 25 kg; 300 mg for 26 to 36 kg; and 400 mg for 37 to 50 kg (126). There are no studies on the effectiveness of gabapentin on spasticity in children.
TIAGABINE
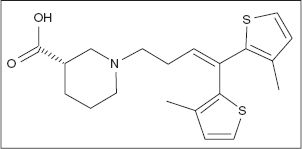
Tiagabine is an anticonvulsant drug that was designed to prevent GABA reuptake into neurons (84). First utilized as an add-on therapy for partial epilepsy (127), more recently it has been utilized as both an adjunctive medication and a monotherapy for complex–partial seizures and generalized tonic–clonic seizures (125,128).
Pharmacokinetics
Tiagabine is 96% absorbed orally in most patients reaching peak serum concentration in 45 minutes in most patients although a delay of up to 3 hours has been reported in those taking other medications that are hepatically metabolized (129). The spectrum of activity and toxicities of tiagabine have been described as similar to those of vigabatrin and gabapentin (114,115,129). Tiagabine is rapidly metabolized via the cytochrome P450 enzyme, and drugs that induce this enzyme will increase its metabolism, including carbamazepine and phenytoin (129–131). However, it has a half-life of only 7 to 9 hours (129,132).
Treatment
Tiagabine is usually initiated at a dose of 4 mg once daily. The dose may be increased by 4 mg at the beginning of the second week and then at weekly intervals by 4 to 8 mg/d, divided in two to four doses, until a clinical response is achieved (133). The maximum dose is 32 mg/d.
Interestingly, there are no studies on the use of tiagabine in adults with spasticity. However, there are two small open-label trials in the use of tiagabine in spastic hypertonia in children (127,134). One small study in 10 subjects indicated an improvement in the Ashworth scores of the children (134). Another study in 14 children who had epilepsy and spastic hypertonia noted an improvement in voluntary motor scores as well (127).
The most frequent adverse effects of tiagabine are dizziness (30%), asthenia (24%), nervousness (12%), tremor (9%), diarrhea (7%), depression (5%), and emotional lability (4%) (4). In one large study, serious adverse effects were described in more than 15% of those exposed to the medication (135). Dosage is usually limited to 6 to 36 mg/d (125). Dosages in the 48 to 60 mg/d were linked to paradoxical nonconvulsive status epilepticus, and hence, excessive dosages should be used with caution in those patients with spastic hypertonia with a documented seizure disorder (136).
VIGABATRIN
Vigabatrin was another attempt to “tailor make” an irreversible inhibitor of GABA-transaminase predominately as an antiepileptic agent (114). By inhibiting the neurotransporters of GABA and increasing the extracellular GABA, it has been demonstrated that this suppresses seizures (114,137). Vigabatrin is broadly licensed in Europe and Latin America. Vigabatirn was approved by the FDA in January of 2009, but its release in the United States had been inhibited by its association to vision loss. The drug has generally been utilized for and now is the only FDA-approved medication as an adjuvant therapy for refractory partial seizures in both adults and children (114,138). Use for spasticity is off-label, and there are no large studies on the use of this drug for spasticity.
Pharmacokinetics
Vigabatrin reaches peak serum concentration within 0.5 to 2 hours (139,140). It is predominately renally cleared (139) and has a plasma half-life of 5 to 8 hours in adults and 12 to 13 hours in elderly persons (4,139). Vigabatrin is not metabolized or significantly protein bound (139,141). The serum levels of liver transaminases are reported to be reduced with vigabatrin, but this has not been associated with hepatic or renal dysfunction (115,139,142). It has been noted to decrease the serum level of phenytoin by 20% to 30% (115,141). The most commonly noted side effects are somnolence/drowsiness (28%), fatigue (28%), dizziness (21%), nystagmus (15%), abnormal vision (11%), agitation (11%), amnesia (10%), depression (10%), and paresthesia (9%) (4,140). Doses usually start at 25 mg/kg/d and increased up to 125 mg/kg/d as needed. One author generally starts most patients on 1000 mg once a day (138), and the drug is generally increased to a maximum of 4 g a day in two divided doses (4,140).
Treatment
Dosing in adults with spastic hypertonia has been in the range of 2 to 3 g/d in two divided doses (141). However, vigabatrin has been of therapeutic value in the treatment of refractory infantile spasms, although these spasms have been linked to seizure activity (143,144). In 43% of the children, there was complete spasm suppression and 95% of the children had a greater than 50% reduction of spasms with initial treatment (143). However, a long-term response was noted in only 75% of children with symptomatic infantile spasms (143).
Vigabatrin can cause psychiatric disturbances, including aggression and psychosis (115,141,145). About half of all the patients experience some adverse effects with vigabatrin. The most common are drowsiness and fatigue, although in children, excitation and agitation occur more frequently. Other CNS-related adverse effects include dizziness, nervousness, irritability, headache, nystagmus, ataxia, paraesthesia, tremor, and impaired concentration. Generally, it is felt that patients with significant psychiatric history should be treated with caution with regard to the use of vigabatrin (128). Toxicology studies have associated vigabatrin with intramyelinic vacuolization or edema in the brains of rodents and dogs (114,115,138). Vigabatrin may need to be withdrawn if visual symptoms develop. Visual field testing should be performed at baseline and during routine follow-up, particularly in children who have visual field deficits or at risk to develop visual field deficits (146–148).
TOPIRAMATE
Topiramate is another antiepileptic drug (AED) for tonic–clonic seizures by blockage of voltage-dependent sodium channels (149), an augmentation of gamma-aminobutyrate acid activity at some subtypes of the GABA-A receptors (4,150,151), and antagonism of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainite subtype of the glutamate receptor affecting Ca channels (151).
Pharmacokinetics
After oral administration, topiramate reaches peak serum concentration in 1 to 4 hours, averaging 2 hours in most patients (152,153), and has an elimination half-life of 21 hours (152). Topiramate is only slightly metabolized, and none of the metabolites is felt to be clinically active (154). It is predominately renally cleared (149,152).
Treatment
Topiramate is usually initiated at doses of 25 to 50 mg/d and increased in increments of 25 to 50 mg/wk (152). It has had limited use in spasticity that has been primarily confined to open-label case series of children with spasticity from Canavan disease, which causes early onset leukoencephalopathy and megalencephaly as well as the infantile spasms of West syndrome, when combined with vigabatrin (4,154,155). It has been reported to be useful in chorea and hemiballismus after stroke in one case report (156). Topiramate’s most significant side effects are somnolence and weight loss (4,152).
CENTRAL ALPHA-ADRENERGIC AGENTS
The monoamines are widely distributed within the CNS. In spastic hypertonia, they appear to modulate sensory, autonomic, and motor functions through facilitation of presynaptic inhibition of spinal afferent inputs (30). The monoamines have an important role as modulators of spinal neuron excitability (157). NE for the spinal pathways is produced in the neurons residing in the brain in the area called the locus ceruleus (30). The mechanism by which noradrenergic pathways modulate spastic hypertonia is predominately by modulating the sensory inputs via presynaptic inhibition of the spinal afferent inputs (30,158). NE also modulates polysynaptic segmental reflexes where NE agonists may restore the vibratory inhibition of spinal reflexes (30,159). NE also has a direct inhibitory effect on interneurons and polysynaptic segmental reflexes (30,160,161). In essence, the central NE modulates spastic hypertonia via central alpha-2 and imidazol type-I adrenergic receptor agonist activity by both reducing the stimulating effect of noxious stimulation (30,162) and modulating the spindle activity directly related to spastic hypertonia (30,163). When the descending pathways from the brainstem to the spinal cord are disrupted, there is a reduction in the NE in the spinal pathways leading to increased spastic hypertonia.
Clinically, these agents have antinociceptive effects that may reduce afferent input into multisynaptic reflex arcs that modulate spasticity and are adjunctive anxiolytics thereby indirectly reducing spasticity as well (164).
CLONIDINE
Clonidine was one of the first central acting alpha-2 and imidazol type-I adrenergic receptor agonists to be utilized in spasticity (30). Clonidine is also an alpha-1 central adrenergic agonist. This effect is what is felt to be attributable to its effects as an antihypertensive medication, which is antagonized by yohimbine (30,165–167). Its effects on spasticity are predominately attributed to its alpha-2 adrenergic receptor effects by presynaptic inhibition of sensory afferents (158,159,167). This is why it is also attributed to be a profound nocioceptive pain reliever (30). Clonidine’s effects on blood pressure appear to be due to the central sympatholytic effect of clonidine. As a result, clonidine has little effect on the blood pressure of people who have complete SCI, but it can lower the blood pressure for normals and those with incomplete injuries (168–170).
Pharmacokinetics
Clonidine is an extremely lipophilic medication with almost uniform distribution whether the medication is delivered via oral (171,172), transdermal (173–175), intravenous (176,177), epidural (178), or rectal delivery (179,180). All reach relatively similar systemic serum levels. Oral clonidine delivered orally reaches peak serum levels in 1 to 1.5 hours and has a serum half-life of 12 to 16 hours (181). Clonidine is 50% metabolized by the liver with the rest renally cleared largely unchanged (181).
Treatment
Clonidine in the oral form has been utilized quite successfully for spastic hypertonia in SCI (30,158,159,168,182). However, all the studies were open-label A–B trials. In patients with stroke, there is one case report (183). Transdermally, clonidine appears to have had a similar effect as orally (184, 185). It should be pointed out that oral clonidine may have profound effects in patients with SCI on autonomic dysreflexia (186) as well as on bladder reflexes, potentially changing the voiding patterns of the patients (169). There are limited pediatric trials using clonidine for spasticity. The predominant side effects are related to its effects on blood pressure. Other side effects include bradycardia, dry mouth, ankle edema, and depression (30,181). Those with a complete high-level SCI may be less likely to have a significant effect on blood pressure (169,170,181).
TIZANIDINE
Tizanidine [5-chloro-4-(2-imidazolin)-2,1,3-benzothiodaiazole hydrochloride] is a centrally acting, selective alpha-2 adrenergic and imidazole agonist that is structurally related to the antihypertensive drug clonidine (30). However, tizanidine has only one tenth to one fiftieth the potency of clonidine in lowering blood pressure or slowing heart rate (187). This is due to the differences in preferential receptor selectivity for alpha-2 receptors rather than alpha-1 adrenergic receptors. Similar to clonidine, tizanidine also has effects on imidazol type I receptors. Tizanidine is active at both segmental spinal and supraspinal levels in both motor and sensory pathways (30). At the spinal segmental level, tizanidine is a presynaptic inhibitor of excitatory amino acid release via the alpha-2 adrenergic and imidazoline receptors (188) and also acts to inhibit polysynaptic reflexes (189). In the brain, tizanidine inhibits firing of the locus ceruleus, thereby inhibiting the normally facilitatory influence of the descending cerebrospinal pathway. Animal models suggest that imidazoline receptors are responsible for the supraspinal effects rather than alpha-2 receptors (188). Tizanidine has no effect on monosynaptic reflexes, such as the standard deep tendon reflex (187). More importantly, tizanidine has no activity at the neuromuscular junction and has no direct effect on skeletal muscle fibers, so it does not cause any muscle weakness (187–192). In some reports, patients actually show apparent increases in muscle strength due to central relaxation of the antagonist muscles (190).
Pharmacokinetics
Tizanidine is well absorbed with 50% to 65% absorption from oral doses and undergoes extensive first-pass metabolism (30). None of the metabolites is pharmacologically active. The half-life of tizanidine is 2.1 to 4.2 hours, with peak plasma concentration reached at 1 hour (193,194). Modified release capsules have a peak serum level of 5.7 hours (195) with 60% of the drug excreted in the urine and 20% recovered in feces (193). An intranasal spray is under development, which has improved bioavailability by bypassing the liver and first-pass metabolism; it also has been shown to have faster time to peak concentration and more predictable plasma concentrations (196). Tizanidine is moderately protein bound (about 30%) and exhibits linear kinetics across the range of usual doses. It has no effect on the hepatic P450 enzyme system (30,193). There is a paradoxical “food effect” that alters the pharmacokinetics of tizanidine. If taken with a meal, the peak plasma concentration is increased by 33%, and the time to reach this peak concentration is reduced by 40 minutes (193). The new modified release capsules somewhat temper this “food effect” (193). The overall absorption of the drug remains unchanged (193). There are drug interactions; particularly with the concomitant use of ciprofloxacin, tizanidine exposure is increased with risks of excessive sedation and hypotension (197,198).
Treatment
Tizanidine has been effectively utilized in randomized clinical trials for spastic hypertonia in SCI, MS, TBI, and stroke utilizing the Ashworth score as one of the outcome measures (190–192,199). In stroke, there is another prominent open-label study also demonstrating significant effectiveness utilizing the Ashworth score as one of its outcome measures (197). A small study has been performed in the pediatric CP population, which showed good reduction in Ashworth score (200), but the medication has been used off-label extensively (201). Dosing should be started at 2 mg at night and increased slowly by 2 mg/d every 2 days to a maximum dosage of 36 mg/d. The effect of tizanidine is clearly dose related (190,202).
The most common side effects of tizanidine are sedation, asthenia, dizziness, and dry mouth (190–194,203) (Table 18.3). Sedation appears to be the most serious side effect in spasticity trials with an incidence of 41% to 46% (190–192). Despite its similarity to clonidine, there is very little hypotension or bradycardia experienced at clinically relevant doses and virtually none in the lower half of the dose range (202). Rebound hypertension can occur with abrupt cessation from higher does (193). Hallucinations and nightmares have been reported, and some gastrointestinal (GI) side effects, notably constipation, can occur in up to 5% of GI patients (193). There is excellent agreement between researchers and clinicians with regard to the type and frequency of adverse drug reactions. Perhaps the most significant issue with the chronic use of tizanidine has been its potential for hepatotoxicity, so liver enzymes should periodically be checked as the dosage is increased (30,193). Adherence to oral spasticity medications has been poor, but is particularly poor with tizanidine (203).
SEROTONERGIC AGENTS
The mechanisms by which serotonin affects spastic hypertonia are not clearly understood and there is likely a complicated interrelation between the afferent and efferent neurons involved. Serotonergic neurons originate in the raphe nuclei of the brainstem and project through the brain and spinal cord (204). It is quite clear that in SCI, the presence of serotonin-containing neurons below the level of injury is an indication of an incomplete lesion (30). However, it is also clear that in serotonin syndrome, there is an increase in tone and spasticity (87,205,206). Serotonin receptors on terminals of the primary afferent and interneurons decreases transmission in the spinal sensory pathway which likely reduces reflex arcs and spasticity (207). Loss of serotonin from descending upper motor neurons in the case of SCI leads to decreased serotonin receptor activity on the interneuron and terminal afferent neurons, which is believed to result in increased reflexes and spasticity (207–209). This would indicate that a serotonin agonist could improve hyperreflexia and spasticity. A study done with zolmitriptan showed decreased H-reflex in incomplete SCI and normal individuals (207); however, this medication is not clinically viable because it cannot be taken regularly due to risk of headaches (210). Alternatively there is evidence that serotonin receptor antagonists and inverse agonists can improve spasticity (208,211), it has been speculated for some time that drugs that can block serotonergic transmission may improve spastic hypertonia (30).
TABLE 18.3
CYPROHEPTADINE
The most profound serotonin blocker utilized in medicine is cyproheptadine, which has been studied in spasticity (211–214) as well as to treat the symptoms of the serotonin syndrome associated with baclofen withdrawal (83). It was initially approved for vascular headaches, anorexia, and hives (4). Studies indicate that constitutively active serotonin receptor blockade has an impact on spasticity, while ligand blockade had no effect; cyproheptadine works through both mechanism (211).
Pharmacokinetics
Besides its profound effects on blocking serotonin, cyproheptadine has profound anticholinergic and antihistaminic effects as well (4,83,215). Dosing is usually at 12 to 20 mg/d in three to four divided doses with dose increases every 3 to 4 days (4,83,214,215). Cyproheptadine reaches peak serum concentration in 6 to 9 hours after oral ingestion and has a serum half-life of 16 hours (216). The drug is 57% conjugated in the liver, and the rest is renally cleared (216,217).
Treatment
Clinical data supporting the use of cyproheptadine are quite limited. It was first reported to decrease ankle clonus in six MS and SCI patients in an open-label study more than 30 years ago (212) and later to improve walking speeds in an open-label study of six SCI patients (213). A larger three-armed open-label trial on 25 SCI patients comparing baclofen, clonidine, and cyproheptadine indicated that on the Ashworth Scale and the pendulum test, cyproheptadine was superior to clonidine and equivalent to baclofen. These data are limited because there was no randomization and no placebo group. More recently, there is a report that cyproheptadine may alleviate some of the effects of intrathecal baclofen withdrawal, indicating that GABA-B receptors do inhibit the release of serotonin and that serotonin is involved in movement disorders (83). The most frequent side effects are somnolence and weight gain with the use of cyproheptadine (30,217).
CANNABINOIDS
Dronabinol (Tetrahydrocannabinol)
Nabilone
Cannabidiol
Cannabis, cannabis extracts, and various synthetic cannabinoids have been touted for years as agents to treat spastic hypertonia. This partly has been the impetus in the United States for various states to legalize the medicinal use of marijuana (4,218). Delta-9-tetrahydrocanibinol (THC) and cannabadiol (CBD) are the two main active ingredients of cannabis, but there are more than 60 psychoactive cannabinoid chemicals occurring naturally. Synthetic versions such as dronabinol, which shares the same structure as delta-9-tetrahydrocannabinol (delta9-THC) and nabilone, which is a synthetic derivative, were developed for nausea (4). Both of these drugs and the cannabis extract have profound antinausea and antianxiety effects; however, there are often side effects related to the psychoactive properties of cannabis, which can make the medications poorly tolerated. Recently a product called nabiximols (Savitex®) has been developed. It is an extract of THC and CBD, and is administered by oral spray (219). The complete mechanisms of action for cannabinoids is not fully understood, but it is known that they function primarily on the cannabinoid receptors (CB-1 and CB-2). CB-1 is primarily found in the CNS and activation of the receptor inhibits neurotransmitter release (220). CB-1 is the receptor that has been implicated in the effect of cannabinoid on spasticity. CB-2 is distributed more widely including cells of the immune system. The effect of THC as a CB-1 agonist has unwanted side effects in treating spasticity including anxiety and psychotic-like symptoms in some patients. The coadministration of CBD reduces these symptoms making a combination medication more tolerable (221). This effect initially led researchers to believe that CBD was a CB-1 antagonist; however, studies have shown that although it has low affinity antagonism in vitro there are no effects on CB-1 in vivo (222). Additionally there may be some anti-inflammatory effects in activation of CB-1 receptors that may improve the progression of MS (223). There has also been an effort to identify an endocannabinoid and modify its degradation, uptake, or production in order to affect the cannabinoid receptor system. Anandamide was identified as a natural ligand for CB-1 and animal research has been done with fatty acid amide hydrolase inhibitors to increase anamdamide activity (224). A mouse model of MS has resulted in decreased spasticity with no cannabis-like side effects (225). This pathway will likely be a future target of human studies.
There has been considerable interest in the use of cannabis, particularly in patients with MS (218).
Pharmacokinetics
Dronabinal reaches peak serum concentration in 1 to 4 hours after ingestion and has a biphasic half-life for the active compounds of 19 to 36 hours (226). The drug is rapidly metabolized into the active compound 11-hydroxy-delta-9-tetrahydrocannabinol and is 10% to 15% renally cleared with the rest cleared in the feces (226,227). Dizziness and somnolence are the most common side effects, although ataxia and memory issues have been reported (226).
Nabilone reaches peak serum concentration in 2 hours and has a half-life of only 2 hours (228). It is extensively metabolized by the liver via the P450 enzyme and is predominately excreted in the feces with 20% to 24% renal clearance (228–230).
Related posts:
 Emerging Technologies in the Management of Upper Motor Neuron Syndromes
Emerging Technologies in the Management of Upper Motor Neuron Syndromes
 Effects of Noninvasive Neuromodulation in Spasticity
Effects of Noninvasive Neuromodulation in Spasticity
 Botulinum Toxin in the Treatment of Upper Limb Spasticity
Botulinum Toxin in the Treatment of Upper Limb Spasticity
 Botulinum Toxin in the Treatment of Lower Limb Spasticity
Botulinum Toxin in the Treatment of Lower Limb Spasticity
 Surgery in the Management of Spasticity
Surgery in the Management of Spasticity
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


