Peroxisomal Disorders
Rebecca S. Wappner
Peroxisomes are small, single membrane–bound, electron-dense, subcellular organelles associated with a growing number of recognized biochemical functions and related disorders. Initially termed microbodies, the name later was changed to peroxisomes when catalase, which reduces hydrogen peroxide to water, and a series of oxidases were found to be localized to this organelle. Peroxisomes are formed by division or budding of preexisting peroxisomes. They then are enlarged and become functional by acquiring newly synthesized membrane and matrix proteins, which contain peroxisomal targeting signals that allow them to be imported into the peroxisomes through receptor-mediated processes.
At least 50 enzymatic reactions are known to occur in peroxisomes. Abnormalities noted in patients with peroxisomal disorders have included catalase deficiency; reduced beta-oxidation of very long-chain fatty acids (VLCFA; carbon chain length greater than 22); reduced biosynthesis of isoprenoids, cholesterol, bile salts, and ether lipids such as plasmalogens; reduced oxidation of phytanic acid, pipecolic acid, and 2-methyl branched-chain fatty acids; and abnormalities in glyoxylate metabolism.
Peroxisomal beta-oxidation of fatty acids differs from that in mitochondria in that the process is not linked to oxidative phosphorylation. Also, the energy produced is not conserved but is dissipated as heat (Fig. 390.1). The peroxisome appears to be the exclusive site for the beta-oxidation of unsaturated VLCFA and plays a major role in the beta-oxidation of monounsaturated long-chain fatty acids (C22:1). Faulty beta-oxidation of VLCFA leads to elevated plasma levels and tissue storage of VLCFA, especially tetracosanoic acid (lignoceric acid; C24:0) and hexacosanoic acid (cerotic acid; C26:0).
Plasmalogens are ether phospholipids that are major constituents of cell membranes, myelin, and platelet-activating factor. Dihydroxyacetone phosphate (DHAP) acyltransferase and alkyl DHAP synthase, peroxisomal enzymes involved with the first two steps of plasmalogen synthesis, have been shown to be deficient in patients with certain peroxisomal disorders.
Peroxisomal mevalonate kinase catalyzes the early steps in the biosynthesis of cholesterol and isoprenoids. Biosynthesis of bile acids (chenodeoxycholic and cholic acids) from cholesterol involves a series of peroxisomal reactions similar to those for beta-oxidation of VLCFA. Faulty biosynthesis leads to the accumulation of bile acid precursors such as dihydroxycholestanoic and trihydroxycholestanoic acids in the serum, urine, and bile.
Phytanic acid, an unusual 20-carbon branched-chain fatty acid mainly of dietary origin, has been noted to be elevated in certain types of the peroxisomal disorders as a result of faulty phytanic acid alpha-oxidation caused by deficient activity of phytanoyl-CoA hydroxylase. Pristanic acid, a metabolite of phytanic acid, accumulates, along with bile salt precursors, in 2-methylacyl-CoA racemase deficiency. Pipecolic acid, an intermediary in the degradation of lysine, also has been noted to be elevated in certain disorders as a result of the deficient activity of pipecolic acid oxidase. Alanine-glyoxylate aminotransferase, which is deficient in hyperoxaluria type 1, is peroxisomal in location.
CLASSIFICATION OF PEROXISOMAL DISORDERS
The peroxisomal disorders presently are classified into two groups. Group 1 includes disorders of peroxisomal biogenesis. Peroxisomal membrane and matrix proteins are not imported into peroxisomes, resulting in generalized peroxisomal dysfunction and the deficient activity of multiple peroxisomal enzymes. The peroxisomal enzymes are transcribed, but remain in the cytosol, where they are degraded. Membranous structures, termed peroxisomal ghosts, may be seen on electron microscopy in tissues from affected patients. These structures may contain some membrane proteins, but they are missing catalase and most other matrix enzymes. Mutations have been shown in PEX (peroxin) genes that encode for peroxisomal membrane proteins, proteins involved with peroxisomal membrane and matrix protein import, and peroxisomal receptors for peroxisomal targeting signals. Considerable variability of
clinical phenotype exists within the same genotype. Some patients have severe forms of the disorders that present in the neonatal period, whereas other forms are milder and present at older ages. Group 1 includes classic and mild Zellweger syndrome, classic and severe neonatal adrenoleukodystrophy (ALD), infantile Refsum disease, hyperpipecolic acidemia, and the rhizomelic form of chondrodysplasia punctata (RCDP).
clinical phenotype exists within the same genotype. Some patients have severe forms of the disorders that present in the neonatal period, whereas other forms are milder and present at older ages. Group 1 includes classic and mild Zellweger syndrome, classic and severe neonatal adrenoleukodystrophy (ALD), infantile Refsum disease, hyperpipecolic acidemia, and the rhizomelic form of chondrodysplasia punctata (RCDP).
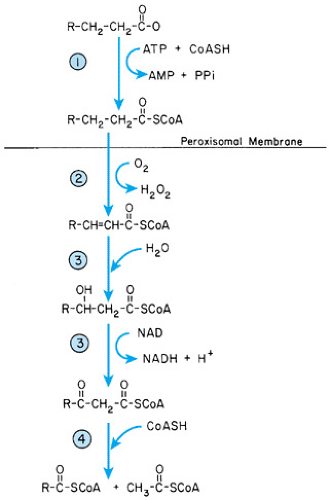 FIGURE 390.1. Peroxisomal beta-oxidation of fatty acids. 1, very long-chain acyl-CoA synthetase; 2, acyl-CoA oxidase; 3, bifunctional enzyme: enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase; 4, 3-oxoacyl-CoA thiolase; AMP, adenosine monophosphate; ATP, adenosine triphosphate; CoASH, uncombined coenzyme A; NAD, nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; PPi, inorganic pyrophosphate. |
TABLE 390.1. BIOCHEMICAL ABNORMALITIES IN PEROXISOMAL DISORDERS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


