Peripheral Neuropathies
Julie Thorne Parke
Involvement of the peripheral nerves may occur in a variety of different disorders, including systemic diseases, infections, and poisonings. In addition, degeneration of the peripheral nerves is a major feature in numerous diseases. Diseases of the peripheral nerve have been classified in several ways. They may be categorized according to type of functional impairment (motor, sensory, autonomic, or mixed), site of pathologic involvement (primary involvement of axon or myelin), clinical course and tempo (acute, subacute, or chronic), presumed etiology, or, in an increasing number of instances, molecular genetics. None of these systems of classification is entirely satisfactory, and combinations of clinical, electrophysiologic, and pathologic features usually are used to determine the etiology. Despite a thorough diagnostic search, the cause of polyneuropathy remains obscure in more than one-half of all cases.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
The term polyneuropathy signifies a generalized disorder of nerve function that usually is symmetric; mononeuropathy implies a disorder of a single peripheral nerve; and mononeuropathy multiplex refers to the dysfunction of multiple single nerves. The clinical signs of a neuropathy reflect the function of the peripheral nerves involved.
The characteristic symptoms of a polyneuropathy are weakness and sensory impairment. The weakness usually is more pronounced distally and often is more severe in the lower extremities than in the upper extremities. A gait disturbance may be an early feature of the disorder. Patients with primarily motor involvement commonly have a high stepping gait that is used to overcome their bilateral foot drop. The distal tendon reflexes usually are absent. In long-standing diseases, such as the hereditary types of neuropathy, wasting of the affected distal musculature may occur, producing an inverted champagne bottle or stork leg appearance of the legs. In a few forms of polyneuropathy, notably Guillain-Barré syndrome (GBS), weakness tends to be proximal and may be attributed mistakenly to a myopathic process. The sensory abnormalities in most neuropathies, similar to the weakness, tend to be distal, becoming gradually less severe in more proximal parts of the limbs. Thus, sensory loss appears to be in a glove-and-stocking distribution on examination. In infants and young children,
profound sensory loss may lead to self-mutilation, involving injury to the insensitive areas. The involvement of motor and sensory neurons may be found in association with various spinocerebellar degenerations, such as Friedreich ataxia, which shows a selective involvement of the posterior column of the spinal cord and results in a marked impairment of proprioceptive and vibratory sensation and less impairment of pain and temperature sensations.
profound sensory loss may lead to self-mutilation, involving injury to the insensitive areas. The involvement of motor and sensory neurons may be found in association with various spinocerebellar degenerations, such as Friedreich ataxia, which shows a selective involvement of the posterior column of the spinal cord and results in a marked impairment of proprioceptive and vibratory sensation and less impairment of pain and temperature sensations.
Pure motor or sensory forms of neuropathy occur, but most disorders cause a combination of motor and sensory symptoms. Predominantly motor polyneuropathies include GBS and the neuropathy of porphyria. Types of neuropathy with severe sensory disturbances but little motor disability include the hereditary sensory neuropathies and some drug-induced neuropathies. If autonomic nerves are affected, abnormalities of pupillary reaction, impaired sweating, impaired bladder and bowel control, and postural hypotension may be noted. Autonomic involvement is a constant feature of the polyneuropathy that is associated with diabetes mellitus and of one form of hereditary sensory neuropathy, the Riley-Day syndrome.
DIAGNOSIS
Several physical signs are helpful in establishing the diagnosis of polyneuropathy. Skeletal deformities, such as pes cavus and hammertoe, are suggestive of long-standing disorders beginning in infancy and usually are caused by hereditary neuropathies (Fig. 407.1). If scoliosis is present, it also is suggestive of a long-standing hereditary neuropathy. Associated retinitis pigmentosa, sensorineural deafness, cerebellar ataxia, or cardiomyopathy suggests a hereditary rather than an acquired disorder. The peripheral nerves usually are normal to palpation, but they may be enlarged in patients with some forms of hypertrophic neuropathy. Enlargement of the peripheral nerves occurs predominantly in patients with demyelinating neuropathies and may be found in those with chronic inflammatory neuropathies and some hereditary neuropathies, such as Charcot-Marie-Tooth disease and Refsum disease.
Electrodiagnostic studies are particularly helpful in diagnosing peripheral neuropathy. Motor and sensory conduction velocities are slowed to varying degrees in patients with most forms of polyneuropathy. In contrast, nerve conduction velocities in patients with anterior horn cell disease or myopathy usually are normal. Nerve conduction velocities may be normal or slowed only slightly in patients with primarily axonal neuropathies, but the amplitude of the compound motor action potential is reduced markedly in these patients. Specialized studies of proximal nerve conduction velocity may be necessary to demonstrate proximal lesions, such as those that occur in GBS. A biopsy of the sural nerve may be useful in making the diagnosis by revealing evidence of either an axonal or a demyelinating process. The specific cause of the neuropathy, however, rarely is established by biopsy alone.
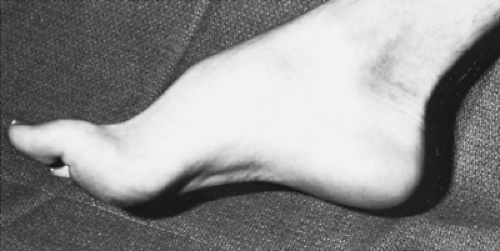 FIGURE 407.1. Pes cavus in a child with a hereditary hypertrophic neuropathy. |
SPECIFIC ETIOLOGIES
Inflammatory Polyradiculoneuropathy (Guillain-Barré Syndrome)
GBS is the most common cause of acute weakness from peripheral nerve involvement. This syndrome is characterized by the acute or subacute development of a polyradiculoneuropathy, usually after an upper respiratory tract infection or an episode of gastroenteritis. Numerous infectious agents, including Epstein-Barr virus, coxsackievirus, influenza viruses, echoviruses, cytomegalovirus, and Mycoplasma pneumoniae, have been associated with the illness. Campylobacter jejuni infection has been associated with GBS, particularly the axonal form. GBS may occur after immunization against rabies. Pathologically, the disorder is characterized by the presence of inflammatory lesions, with segmental demyelination scattered throughout the peripheral nervous system. The most severely involved segments are the rootlets and the proximal portions of the peripheral nerves.
Much evidence supports an immunologic basis for this disease. The neuropathologic and clinical features are similar to those of an experimental condition known as experimental allergic neuritis, which is induced in animals through the injection of Freund adjuvant into peripheral nerve tissue. Experimental allergic neuritis can be transferred passively between animals by sensitized lymphocytes but not by serum, thus suggesting that experimental allergic neuritis is mediated by a delayed hypersensitivity mechanism. The prevailing opinion is that demyelination in GBS is secondary to a cell-mediated immune response that is directed against a component of peripheral myelin. Humoral immunity also has been found to be altered in patients with GBS, and it may contribute to the pathogenesis of the disorder.
Clinical symptoms typically manifest after an antecedent infection, following a latent period that varies in length from several days to several weeks. The most common initial symptoms are numbness and paresthesias of the hands and feet, followed by progressive weakness involving all four extremities. Motor impairment usually begins in the lower extremities and progresses in an ascending pattern to involve the upper extremities, trunk, and cranial nerves. A descending pattern of weakness also has been observed. Occasionally, the onset is abrupt, with simultaneous involvement of all extremities. The weakness usually is symmetric, although minor differences between the sides may occur.
A spectrum of motor involvement, varying from mild weakness to a complete flaccid quadriplegia, occurs. Muscle stretch reflexes are markedly reduced or absent. Involvement of the cranial nerves is seen commonly, with facial diplegia occurring in 50% of patients. Lower cranial nerve dysfunction may give rise to dysarthria and difficulty in swallowing and coughing. Significant respiratory muscle weakness occurs in 20% of patients and may necessitate artificial ventilation. Sensory symptoms are much less prominent than is weakness, but a distal sensory loss, particularly involving proprioception and vibratory sensation, may be present.
The autonomic nervous system is involved frequently, with episodes of paroxysmal hypertension or hypotension, tachycardia or bradycardia, facial flushing, and sweating abnormalities. Bowel and bladder functions may be impaired early in the course of the disease, but sphincter dysfunction usually is short-lived. The neurologic symptoms evolve fairly rapidly over the
course of the first few days, with maximum disability reached within 1 week in most cases. A stable period of 1 to 3 weeks occurs, after which recovery begins. The recovery may be rapid, taking place in 6 to 8 weeks, or it may be slow, lasting many months.
course of the first few days, with maximum disability reached within 1 week in most cases. A stable period of 1 to 3 weeks occurs, after which recovery begins. The recovery may be rapid, taking place in 6 to 8 weeks, or it may be slow, lasting many months.
TABLE 407.1. CRITERIA FOR DIAGNOSIS OF GUILLAIN-BARRÉ SYNDROME | ||
|---|---|---|
|
Many patients with GBS have some variation in clinical presentation or laboratory test results. The currently accepted criteria for the diagnosis of this syndrome are listed in Table 407.1. Several variants of GBS are recognized; the most common one occurring in childhood is a syndrome of acute external ophthalmoplegia, ataxia, and areflexia known as the Miller-Fisher syndrome. The ophthalmoplegia often is bilateral and may be complete, with pupillary involvement. The course usually is benign, with recovery taking place within 3 to 6 months. More recently, a severe acute sensory variant has been described.
The most important laboratory finding in patients with GBS is an elevated cerebrospinal fluid (CSF) protein content without a pleocytosis (albuminocytologic disproportion). The total CSF protein level may be normal in the early stages of the illness, but it is elevated in almost all patients after an interval of several days. The protein content continues to increase after the disease stabilizes, reaching a peak 2 to 4 weeks after the onset of the disease and ranging from 45 to 800 mg/dL.
Electrophysiologic studies are helpful in diagnosing GBS, with abnormalities of motor and sensory conduction occurring in 90% of patients. Characteristic electrodiagnostic features include marked slowing of conduction velocities, prolonged distal latencies, and dispersion of the evoked responses. Proximal nerve conduction, which characteristically is slow and can be measured by studying the latency of the F response, may be the only abnormal electrophysiologic finding in the early stages of the disease. In later stages of the disease, electromyographic studies may show denervation potentials indicating axonal damage, which is associated with a poor prognosis for complete recovery.
The treatment of GBS is largely supportive. Careful monitoring of respiratory function is important during the early stages of the illness to prevent death as a result of respiratory failure. Elective intubation and mechanical ventilation should be used aggressively in patients with any evidence of respiratory compromise because respiratory failure may occur abruptly if they become fatigued. Good nursing care and physiotherapy are important in severely affected patients. Most children with GBS recover completely, although the convalescence may be prolonged. Plasmapheresis and intravenous immunoglobulin have been shown to be beneficial both in shortening the length of the illness and in lessening the severity of the associated long-term disability.
Numerous entities may produce a clinical picture similar to that of GBS. The ascending form of acute transverse myelitis and early cord compression may be difficult to distinguish from GBS initially. The presence of pyramidal tract signs, a clear sensory level, and persistent sphincter disturbances support involvement of the spinal cord rather than the root and peripheral nerve. Acute paralytic poliomyelitis may present with weakness simulating GBS, but generally more systemic symptoms, more marked meningeal signs, and a cellular response in the CSF are present. Uncommon conditions that may cause acute symmetric weakness include porphyria, diphtheritic polyneuropathy, heavy-metal intoxication, systemic lupus erythematosus, periodic paralysis, tick paralysis, rabies, and botulism.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


