INTRODUCTION
Pediatric rehabilitation encompasses a wide variety of conditions, from neurologic disorders to musculoskeletal abnormalities that affect growth and development. Developmental delay, specifically in gross and fine motor activity, is a common concern of parents and caregivers; thus an understanding of normal growth and developmental patterns is essential (Table 20–1). Causes of developmental dysfunction in motor and cognitive skills range from congenital and genetic abnormalities to acquired illnesses and injuries of the neurologic and musculoskeletal systems. A thorough history and examination is key to diagnosis and management. Goals for habilitation or rehabilitation will depend on the child’s age and development.
GENERAL CONSIDERATIONS IN PEDIATRIC EVALUATION
In the evaluation of pediatric patients, the importance of the chief complaint and reason for the visit cannot be overemphasized. Traditionally part of the basic history, this component is sometimes omitted or inferred by time-pressed providers. Of similar importance are the parents’, caregiver’s, or patient’s goals and expectations, which will shape the direction of later care and management.
In addition to obtaining past medical and surgical histories, the examiner should explore the patient’s birth history. Information about prenatal care, maternal illness or conditions, medications, mode of delivery, age of gestation at delivery, and any perinatal and postnatal complications (including treatment course in the neonatal intensive care unit [NICU]) can provide important clues that may help in the differential diagnosis. Queries should be made regarding developmental milestones and functional abilities, including the age at which milestones were achieved, delays, and any skills that were lost or have regressed (see Table 20–1). Relevant information from the family, personal, and social history, including concerns relating to school performance and social interactions, medications and allergies, and immunizations, should be noted.
| Age | Gross Motor | Fine Motor | Visual Perception | Language | Social–Emotional Play |
|---|---|---|---|---|---|
| Newborn | Arms and legs flexed Poor head control | Hands fisted Involuntary grasp reflex | Can fixate on a face at 8–15 inches Visual acuity 20/400 | Startles or widens eyes to sound Variation in crying | Fixates on a face in preference to other objects |
| 2 months | Head lag on pull to sit Lifts head in prone Head erect when held upright | Grasp reflex disappears Hands open and relaxed Hands to midline Holds objects put in hand | Can track horizontally and vertically | Coos and laughs Vocalizes with vowel sounds | Social smile Responds |
| 3 months | No head lag on pull to sit Can lift chest when prone | Reaches and swipes a toy | Can track a ring in a circular motion Stares at own hand | Coos and laughs Vocalizes with vowel sounds | Interested in image in mirror, smiles; playful Laughs at active stimuli |
| 4 months | Rolls over back to side | Voluntary grasp | Localizes bull’s eye in near and far position | Squeals | (+) Gaze monitoring |
| 6 months | Can sit with support Rolls over front to back | Raking motion Transfers object from hand to hand | Can look for a dropped spoon Pulls a cord to obtain a disc | Babbles with consonant sounds Turns to orient to name | Basic emotions emerge: happiness, interest, surprise, fear, anger, sadness, and disgust |
| 9 months | Sits without support Can sit from supine Pulls to stand Crawl Cruises along furniture | Radial digital grasp Immature pincer grasp Uses forefinger to poke or roll an object | Can look for a hidden object Turns cup right side up | Says “mama,” “dada” nonspecifically polysyllabic babbling (+) Joint attention | (+) Stranger anxiety Engages in back-and-forth play and peek-a-boo Attachment to preferred caregiver |
| 11 months | Stands alone Walks with hands held | Puts small objects in a cup | Makes object association | Says “mama,” “dada” specifically Gives toy with gesture | Shows or offers a toy to adult |
| 12 months | Stands alone Can take a few steps | Mature pincer grasp Turns pages in a book | Demonstrates object permanence Attends to a picture | Says at least 1 word clearly Can identify objects | Points to an object to obtain it Attachment forms Symbolic play |
| 15 months | Begins to walk alone Gait is wide-based | Spontaneous scribbles Builds a tower of 2 blocks | Looks for a toy that was displaced Can put a circular shape in a puzzle | Says 2 words in addition to “mama” and “dada” Gives a toy on request without gesture Combines jargon and gesture | Greets people with “hi” Recognizes image of self in mirror |
| 18 months | Can climb into an adult chair Begins to run Walks up stairs with help | Builds a tower of 3 blocks Imitates a vertical line | Deferred imitation Can put 4 shapes in a formboard | Uses > 5 words; follows simple instructions Can identify 4 body parts | Points to share an experience Uses the word “no” |
| 24 months | Climbs up and down stairs Walks on tiptoes Jumps in place Runs well | Imitates vertical and circular strokes Can feed self with a spoon Puts on simple clothes | Can match 3 objects without naming Can nest 4 cups | Uses 100–200 words, 2-word phrases Speech is 50% intelligible Uses personal pronouns: “me,” “mine,” “I” Identifies 6 body parts Speaks in present tense | Responds to correction Self-conscious emotions emerge Empathy appears Parallel play |
| 30 months | Jumps from a step Hops 1–3 times on same foot | Builds a tower of 8 blocks Zips and unzips | Can sort items Can match pictures | Understands prepositions | Substitutes objects for another |
| 36 months | Can pedal a tricycle Hops 4–6 times on same foot | Copies a circle Fastens and unfastens large buttons Uses a spoon effectively Cuts paper with scissors | Demonstrates memory for a picture Can match a shape by size and color Understands spatial concepts (bigger, smaller) | Speaks in 3–4 word sentences Speech 75% intelligible Uses plurals Uses “what” and “who” questions Can identify 2 colors | Complementary role playing Increased fantasy play (superhero) Good and bad themes predominate |
| 4 years | Walks up and down steps, alternates feet Broad jump Hops on same foot | Copies a cross, square Holds a crayon well Uses a fork | Discriminates left and right | Speech is 100% intelligible Identifies gender Uses “why” questions | Cooperative play Understands the perspective of others |
| 5 years | Single-leg stance for 10 seconds Can skip | Copies triangle Prints some letters | Increased spatial awareness | Defines simple vocabulary | Increased pretend play |
| 6 years | Walks securely on a balance beam | Can tie shoes Mature tripod pencil grasp Copies a diamond | (+) Memory for complex spatial forms | Reading is by word recognition Can repeat complex sentences | Play involves games with rules Moral self continues to emerge |
The physical and neurologic examination of a child, especially an infant or toddler, can be challenging and may be limited by the patient’s inability to cooperate or communicate with the examiner. The examination can be facilitated by developing a rapport with the parent or caregiver and child before touching the child, and, particularly with infants and young children, by incorporating play and use of toys, music, and songs in the assessment. The examination starts by simply observing the child, noting any dysmorphic features or gross abnormalities such as head or facial asymmetry, neck and limb posturing, or limited movements and chest deformities (pectus carinatum or excavatum). Patterns of breathing and the presence of a tracheostomy or a feeding tube should be noted.
In the extremities, the examiner may notice limb-length discrepancy, hemihypertrophy, or uneven skinfolds, especially at the thighs, which may be indicative of hip dysplasia. Active and passive range of motions are tested and any limitation of the joint or contractures, whether fixed or flexible, are noted. Common foot alignment abnormalities with bony deformities include talipes equinovarus (clubfoot), pes planus or pes planovalgus (flat feet), and pes cavus. Skin tags, hypopigmentation or hyperpigmentations, whorl patterns, birth marks, or hirsutism should be noted as these may be associated with a genetic syndrome. Table 20–2 lists distinctive clinical findings associated with common genetic syndromes. In examining the back and spine, the examiner should observe for presence of a sacral dimple or sinus, hair tuft, or abnormal spinal curvatures.
| Syndrome | Genetics/Inheritance Pattern | Clinical Findings |
|---|---|---|
| Down syndrome | Trisomy 21 | Hypotonia, flat facies, slanted palpebral fissures, small ears, mental deficiency, endocardial cushion defect (40%), short neck, hyperflexible joints, high risk for C1–2 subluxation |
| Edward syndrome | Trisomy 18 | Clenched hand with overlapping fingers, short sternum, low-arched dermal ridge, patterning on fingertips, feeble cry, ventricular or atrial septal defect, hypotonia and hypoplasia of skeletal muscles |
| Patau syndrome | Trisomy 13 | Holoprosencephaly, microcephaly, severe mental retardation, polydactyly, narrow convex fingernails, prominent (rocker-bottom) heel, cleft lip or palate, cardiac abnormality |
| Klippel-Feil syndrome | Mutation in GDF6 and GDF3; autosomal dominant | Fusion of any cervical vertebra 2–7, restricted neck range of motion, short neck, low hair line, spina bifida, scoliosis |
| Cornelia De Lange syndrome | Autosomal dominant, mutation in Nipped-B homolog | Synophrys (unibrow); hirsutism; thin, downturning upper lip; micromelia or limb deficiency; low-pitched cry; mental retardation |
| Turner syndrome | 45 XO | Short stature, lymphedema, webbed neck from cystic hygroma in infancy, coarctation of the aorta, bicuspid aortic valve, horseshoe kidney, attention deficit hyperactivity disorder, amenorrhea |
| Noonan syndrome | Autosomal dominant | Webbed neck, pectus excavatum, cryptorchidism, pulmonic stenosis, cardiac defects, short stature, scoliosis |
| Prader-Willi syndrome | Deletion of paternal chromosome 15 | Hypotonia, obesity, small hands and feet, scoliosis, excessive appetite, mental retardation |
| Angelman (“happy puppet”) syndrome | Deletion of maternal chromosome 15 | “Puppet-like” gait, ataxia, jerky arm movements, paroxysms of laughter, developmental delay, speech impairment |
| VATERR association | Unknown | Vertebral anomalies, anal atresia, tracheo-esophageal fistula, radial dysplasia, renal anomaly |
| Ataxia–telangiectasia syndrome | Autosomal recessive, ATM gene mutation, chromosome 11 | Progressive ataxia, telangiectasia, dysarthria, lymphopenia, immune deficit |
| Rett syndrome | MeCP2 mutation | Regression of milestones, hand-wringing or handwashing, dystonia, breath-holding, unsteady gait, severe constipation |
| Friedreich’s ataxia | Autosomal recessive, abnormal frataxin protein (trinucleotide repeat) | Progressive ataxic gait, dysarthria, muscle weakness, decreased proprioception and vibration sense, cardiomyopathy |
As part of the neurologic assessment, the child’s level of alertness, attentiveness, and social interaction, including speech/language and communication, are observed with reference to age-appropriate expectations. Cranial nerve function is assessed for deficits and abnormalities. The motor assessment includes evaluation of muscle tone, strength, and the presence of abnormal or involuntary movements. If formal manual muscle testing cannot be accurately performed (eg, in younger patients), a description of movements or ability to perform tasks may suffice. If the patient is able to walk, any gait deviations, abnormal or compensatory mechanisms involving limb posture, trunk alignment, step and stride lengths, base of support, and balance should be noted. Sensory and cerebellar functions are also assessed. Reflex testing includes muscle stretch responses; the presence of primitive or infantile reflexes, and their abnormal persistence, should be noted (Table 20–3).
CEREBRAL PALSY
ESSENTIALS OF DIAGNOSIS
A group of disorders affecting movement and posture, and causing activity limitation.
Attributed to nonprogressive injury and disturbances in the developing fetal or infant brain.
Often accompanied by disturbances of sensation, perception, cognition, communication, and behavior; epilepsy; and secondary musculoskeletal problems.
Cerebral palsy is the most common cause of motor disability affecting children. In the United States, the Centers for Disease Control and Prevention estimates that 3.3 per 1000 children have cerebral palsy. This is similar to European data of 1.5–3 per 1000 live births. The incidence has increased over time—a fact that may be attributed to technical and medical advances, which have increased the survival of low-birth-weight and premature infants. Although there is no racial or ethnic predilection, the incidence of cerebral palsy has been correlated with low socioeconomic status.
Cerebral palsy is caused by injury or developmental disturbances to the immature brain. While the actual etiology may not always be well understood, certain risk factors may contribute to its occurrence. These include prematurity, infection, inflammation, trauma, and coagulation disorders leading to intrauterine or perinatal strokes.
The most common risk factor for cerebral palsy is prematurity. Infants born at or before 28 weeks of gestation, with low or very low birth weights (1000–1499 g) are at particularly high risk given the potential for multiple medical complications, including intraventricular hemorrhage. Maternal risk factors include infections such as chorioamnionitis, endocrine disorders (especially thyroid disease), fever during labor, multiple births, and placental insufficiency or abnormality. Intrauterine infections and postnatal sepsis, meningitis and encephalitis, asphyxia, hypoxic ischemic encephalopathy, and toxins are also important causes. Kernicterus (bilirubin deposition at the basal ganglia due to severe jaundice) is rarely encountered in the United States but may be seen in developing nations. Traumatic brain injury, including abusive head trauma (“shaken baby” syndrome), is also a significant cause. Whether structural brain anomalies such as lissencephaly or holoprosencephaly should be classified under cerebral palsy is debated, as both are congenital disorders rather than caused by injury. However, clinical manifestations and management for both conditions are similar to cerebral palsy.
The classification of cerebral palsy has evolved since the condition first became a focus of medical research by Dr John Little in the 19th century. Historically, cerebral palsy has been grouped into four main motor syndrome categories: spastic, dyskinetic, ataxic–hypotonic, and mixed type. Spasticity, as defined by the Task Force on Childhood Motor Disorders, is hypertonia manifested as resistance to a rapid stretch or change in joint angle. Dystonia is a movement disorder that causes twisting and repetitive, abnormal postures, or both. Although the spastic pattern occurs most commonly, a mix of spasticity and dystonia is also seen. Children whose movements are predominantly dystonic tend to have lesions in the deep brain structures, including the basal ganglia and thalamus. The ataxic–hypotonic form of cerebral palsy is the least common.
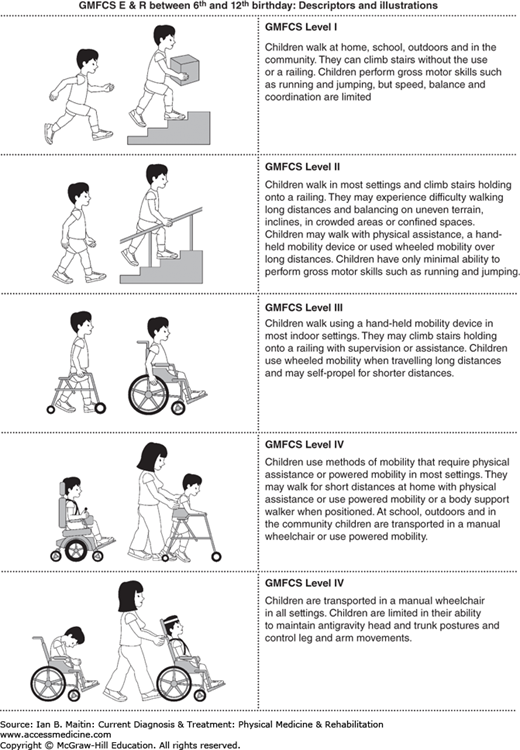
Owing to the clinical complexity and diversity of cerebral palsy, there has been a push for development of classification systems that would better reflect the underlying pathology and would improve standardization of treatment. In 1997, Palisano and colleagues established a more reliable quantitative classification of gross motor function that places more focus on what the child is capable of doing with regard to sitting, walking, and wheeled mobility. The Gross Motor Function Classification Scale (GMFCS) is now widely used to help guide intervention and management (Figure 20–1). In 2004, the International Workshop on the Definition and Classification of Cerebral Palsy recommended a more comprehensive approach to the specification of motor abnormalities, including the nature and type of the movement disorder, functional motor abilities, accompanying impairments, anatomic and neuroimaging findings, and causation and timing of injury.
Numerous illnesses cause motor deficits that can mimic cerebral palsy. Diagnosis is usually made between the ages of 2 and 5 years as notable persistent developmental delay, weakness, abnormal muscle tightness, or the child’s inability to walk prompts further investigation. A detailed history and physical examination are essential in narrowing the diagnosis. Along with the past medical history, the examiner should carefully review the birth history (including NICU course), family history, achievement of developmental milestones, functional impairments, and early hand preference (in children younger than age 2). Parental or caregiver concerns about weakness, abnormal muscle tightness, poor head or neck control, and unusual means of mobility (eg, bunny hopping and combat or commando crawling) should be noted.
A thorough neurologic examination should begin with observation of the child’s movement and posture. Muscle tone is assessed by gently moving the joints through their appropriate range of motion and evaluating the degree of resistance. The Modified Ashworth and Tardieu scales are useful tools in evaluating spasticity. Range of motion of each joint is evaluated for muscle tightness or contractures. Along with hypertonia, children with cerebral palsy may present with loss of selective motor control, muscle weakness, and impaired balance.
Children with hypotonic cerebral palsy usually have difficulty maintaining a sitting position owing to weakness of the neck and trunk and a frog-leg posturing of the legs, whereas those with spastic quadriplegia have hips that cross at the midline or extensor posturing of the legs. The arms may be in a flexor pattern posture with shoulders adducted and internally rotated, elbows flexed, forearm pronated, and hands fisted. Hemiplegic children show early hand preference and decreased ability for bilateral motor tasks. Walking onset may be delayed but almost all children eventually become ambulatory, developing an abnormal gait pattern similar to that of a patient with stroke, and manifesting a spastic flexor pattern synergy of the upper limb and extensor pattern in the lower limb.
Children with diplegic cerebral palsy tend to have better trunk control in sitting than those with quadriplegia, but have impaired dynamic balance and may need support or an assistive device to stand and walk. They tend to ambulate with a scissoring and tiptoe (equinus) pattern because of hip adductor and ankle plantarflexor spasticity. A crouched gait pattern is seen in older children as a result of lower extremity weakness and progressive contractures.
In dystonic or dyskinetic cerebral palsy, muscle tone may be variable or fluctuating and there is sustained muscle contraction, with twisting or repetitive movements usually triggered or pronounced with voluntary effort. Children with pure dystonic cerebral palsy rarely have joint contractures. Some patients may have other types of abnormal movements, such as chorea and athetosis.
Muscle stretch reflexes are usually hyperreflexive, and clonus may be elicited, especially at the ankles and knees. Persistence of primitive infantile reflexes (eg, Moro, asymmetric tonic neck reflex, and palmar and plantar grasp) past 6 months of age should raise red flags for cortical developmental abnormality. The emergence of parachute, equilibrium, or righting reflexes may be delayed or impaired, and sometimes these postural or protective reactions fail to develop at all.
Although the brain disorder or injury is one that is static, secondary musculoskeletal problems are likely to evolve and progress as the child grows. Equinus deformity resulting from spasticity and contracture of the gastrocnemius–soleus complex is a common finding. Equinovarus deformity has the additional varus or inversion component and is caused by tightness of the posterior tibialis muscle. Other foot deformities include equinoplanovalgus and equinocavovarus.
At the knee, flexion contractures occur due to spasticity and tightness of the hamstring muscles. Such contractures may result in a crouch gait pattern among ambulatory patients and, in severe cases, lead to posterior pelvic tilt and decreased lumbar lordosis in the seated position. Patella alta (high-riding patella) can occur due to increased muscle pull of the rectus femoris muscle and may result in a stiff-knee gait, characterized by difficulty in flexing the knee during swing phase. Genu valgum is often associated with significant femoral anteversion and hip internal rotation.
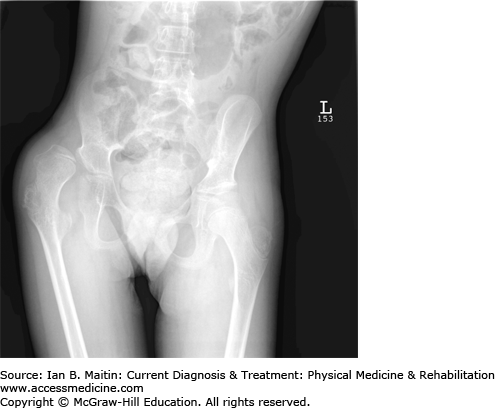
Special attention should be given to evaluating the patient for hip displacement, which is caused by spasticity of the hip adductors and underdevelopment of the acetabulum. Hip subluxation is defined as a migration percentage of greater than 30% on radiographic examination; this may eventually lead to frank dislocation (Figure 20–2). Limited hip abduction and external rotation, along with a positive Galeazzi’s sign (ie, the weaker leg in hemiplegic patients is shorter) and pain on passive range of motion at the hips should raise suspicion of hip pathology and prompt an imaging study. Up to one third of children with cerebral palsy develop hip displacement, with the severity of displacement directly related to the GMFCS level (most severe in levels IV and V). Windswept deformity occurs when one hip is adducted and internally rotated, while the opposite side is abducted and externally rotated. Hip surveillance is recommended as early as age 2, followed by serial hip radiographs, tone management, and orthopedic intervention as necessary, with the goal of preventing hip dislocation, which can lead to pain, decreased mobility, and difficulty in seating and positioning. Other common rotational limb abnormalities include femoral anteversion and excessive internal tibial torsion.
Neuromuscular scoliosis is a common problem in children with cerebral palsy, with a reported incidence of up to 67%. Most cases occur in patients classified as GMFCS levels IV and V. Severe spinal curvatures, including kyphosis, can impede proper positioning and seating, cause skin irritation or breakdown, and compromise respiratory function.
Osteopenia is also common, placing children with cerebral palsy at increased risk for long bone fractures with minimal trauma. No clear guidelines are available with regard to treatment. Most clinicians recommend vitamin D and calcium supplementation while encouraging weight-bearing activities as much as possible for bone health. Bisphosphonate treatments are reserved for patients who have experienced multiple fractures or those with vertebral compression fractures.
Although seizure occurrence varies among the different cerebral palsy groups, children with hemiplegia or those with severe motor impairments (ie, quadriplegia) often have epilepsy and related disorders.
Sensory assessment is difficult, particularly among infants and children with cognitive impairment. Those with hemiplegic cerebral palsy may have concomitant hemisensory loss or impairment to light touch and pain. Visual disturbances and ophthalmologic abnormalities are frequently noted in children with cerebral palsy. Strabismus is the most common finding. Other abnormalities include cortical visual impairment, amblyopia, convergence disorder, refraction errors, and astigmatism. Hearing loss or impairment is relatively rare but may be noted among patients with a concomitant genetic syndrome or abnormality.
Dysarthria, dysphagia, and sialorrhea may lead to communication difficulties, decreased caloric intake, and aspiration pneumonia. A swallow function test is usually warranted to help guide feeding and prevent aspiration (see Chapter 38), and referral to a feeding therapist is recommended for assistance with strategies and oromotor strengthening.
Speech and language delay is also common and tends to be more severe among children with quadriplegic cerebral palsy. Verbal skills may be significantly impaired in a child with normal cognition; thus, other forms of communication (eg, sign language or use of an augmentative communication device) should be explored.
Cognition level varies among children with cerebral palsy, but mental retardation is more commonly associated with the spastic quadriplegic pattern. Patients with hemiplegic and diplegic motor dysfunction may have normal intelligence or some degree of learning disability requiring special education services with an individualized education plan (IEP) or resource classes. It is important to note, however, that severity of motor and expressive language impairments does not equate with mental retardation. Cognition level can be normal or even above average, especially in those with lesions involving extrapyramidal structures (eg, ataxic, dystonic, or athetoid), and an accurate assessment of cognitive skills is needed to ensure appropriate educational and vocational support. Psychological, behavioral, and emotional problems include attention deficit/hyperactivity disorder (ADHD), impulsivity, aggression, passivity, and low self-esteem. Some children may have autistic or pervasive disorder characteristics.
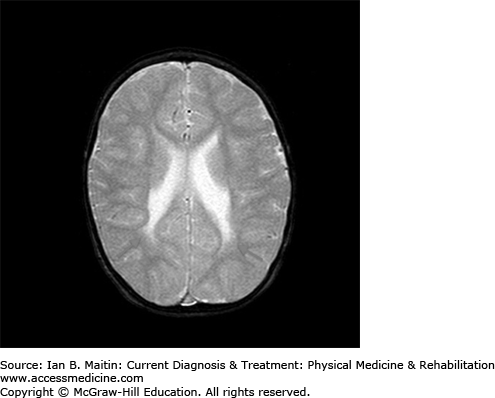
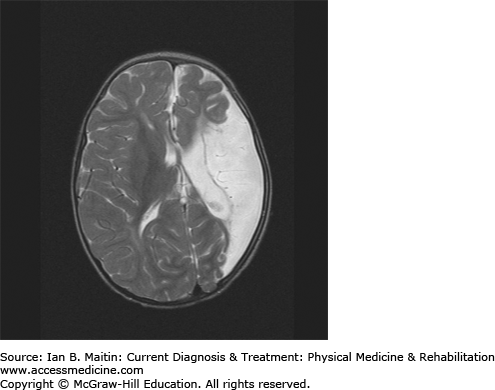
Neuroimaging is important in identifying lesions or abnormalities that support the diagnosis of cerebral palsy, while elucidating the pathophysiology behind the clinical presentation. Magnetic resonance imaging (MRI) is the imaging study of choice as it may help in clarifying the onset or time of injury. Periventricular leukomalacia is a classic finding that commonly occurs as a result of intraventricular hemorrhage in premature infants (Figure 20–3). Among term infants, neuroimaging findings are often normal and no specific lesions may be identified. Infants with intrauterine infarction tend to show focal ischemic brain changes within a single or multiple vascular distributions. Unilateral middle cerebral artery infarction (Figure 20–4) is seen in approximately one third of children with hemiplegic cerebral palsy. A coagulation panel should be obtained to check for a possible hematologic disorder contributing to the occurrence of stroke in a child with cerebral palsy.
In making the diagnosis of cerebral palsy it is important to establish, through the history and physical examination, that there has been no regression or loss of previously acquired skills, and that findings do not suggest a progressive or degenerative central nervous disorder. Functional impairments may worsen over time in children with cerebral palsy as a result of secondary musculoskeletal complications from abnormal muscle tone and weakness, but no new neurologic findings should be noted on subsequent examinations. Migrational or developmental anomalies such as holoprosencephaly and lissencephaly (smooth brain) may be detected and often are associated with dysmorphic features or other congenital anomalies. In these cases, genetic testing may be warranted. Important differential diagnoses for patients evincing the spastic diplegic pattern include neuromuscular disorders and myopathy (especially for children with hypotonic-type findings), and Rett syndrome. Hereditary spastic paraparesis and spinal cord lesions such as tumor or tethered cord may also mimic cerebral palsy, and imaging of the spine may be warranted to rule out these conditions, especially when symptoms seem progressive or the history does not reveal risk factors for cerebral palsy. The workup to identify metabolic causes includes, but is not limited to, a basic metabolic panel; plasma amino acid analysis; arterial acid–base levels; ammonia, lactate, pyruvate, and bilirubin levels; and urinalysis (organic acid analysis). Lumbar puncture to obtain cerebrospinal fluid for evaluation of neurotransmitter disorders or glucose transporter deficiency may be requested, as these conditions are highly associated with seizures and movement disorders.
The most favorable habilitation or rehabilitation results are achieved when goals are realistic. Striving to achieve outcomes that are beyond the patient’s reach can ultimately leave both child and family discouraged. Frequent review of the patient’s progress is key, while simultaneously inviting feedback from primary caregivers throughout the rehabilitation process. The rehabilitation goals for a child with cerebral palsy are unique to each patient and ideally should combine the goals of both the caregivers and the rehabilitation team.
There is significant statistical evidence to verify improved functional mobility through muscle strengthening in patients with cerebral palsy whose muscle weakness impaired their functional mobility but whose muscles were still able to generate enough volitional movement to be trained. The National Guideline Clearinghouse has published a best evidence statement on strengthening for individuals with cerebral palsy, aged 4–20 years, who demonstrate muscle weakness. It recommends strength training in small numbers of repetitions until fatigued, with rest periods between exercises. Strength training should be task-specific and tailored to the upper or lower extremities, or both, as indicated. Several techniques can be used to enhance developmental stimulation and promote functional improvements.
Aquatic therapy can be helpful in achieving goals related to increased endurance by forming more efficient movement patterns and assisting in respiration and phonation. Warm water relaxes taut muscles, leading to a decrease in tone and, hence, to the development of more efficient movement patterns. In addition, hydrostatic pressure activates sensory receptors and increases external lung pressure, thereby facilitating breathing and speaking.
Constraint-induced movement therapy (CIMT) and bimanual training may be used to improve upper limb and fine motor function in patients with hemiplegic cerebral palsy. CIMT may be used to help facilitate use and function of the plegic and neglected extremity. Studies comparing CIMT with bimanual training have been inconclusive in determining whether one is more advantageous than the other; improvements in limb use and function, as well as quality of life, are reported with either technique, as well as both in combination. Other therapeutic modalities include neuromuscular electrical stimulation, biofeedback, and therapeutic horseback riding or hippotherapy, and partial body weight–supported treadmill training.
Oral aversion is often the initial problem in feeding, especially among ill infants who have been receiving long-term parenteral or enteral tube feedings. Oromotor stimulation and desensitization techniques can be applied. Children with oromotor weakness and dysphagia should be evaluated by a feeding therapist. A speech and language pathologist can also address issues relating to swallowing, as well as speech and language deficits, including verbal and nonverbal communication. (Chapter 38 covers this evaluation in detail.) With advances in technology, several adaptive computer devices are now being utilized as adjuncts for communication.
For patients with moderate to severe dysphagia, placement of a nasogastric or gastrostomy tube may be indicated to ensure safe and appropriate nutrition. Gastroesophageal reflux (GER) is highly prevalent, especially among premature infants, and should be properly managed through a combination of positioning and medications to decrease the gastric pH and prevent vomiting. A Nissen fundoplication may be performed upon placement of a gastrostomy tube in children with severe GER.
Children with cerebral palsy often have difficulty managing oral secretions due to oromotor weakness and poor swallowing. This may lead to anterior drooling (sialorrhea) or posterior drooling and aspiration. Management options include anticholinergic medications such as glycopyrrolate, scopolamine patch, and atropine drops. Botulinum toxin can also be injected into the salivary glands, and more definitive surgical intervention can be performed (eg, salivary duct ligation or excision).
For a flexible joint contracture, a cast (soft, reinforced soft, or bivalve plaster or fiberglass) is applied for 1–4 weeks and changed every 7–10 days. This cycle continues while the joint range of motion, gait, and other physical findings are reassessed until the ankle or knee range of motion goals are achieved. The goals of this extensive therapy may include increasing the passive range of motion of ankle dorsiflexion or knee extension, or both, in order to improve brace fit, increase range of motion during daily stretching routines, advance overall function, or delay more invasive procedures.
Various orthotic devices are commonly recommended for children with cerebral palsy. Goals for application include providing stability, preventing contractures, improving range of motion, improving positioning, and promoting better alignment of the foot–ankle in standing and walking. The most common lower limb orthosis is the ankle–foot orthosis (AFO). This can be solid or articulated depending on the goal and the patient’s level of function. Generally for children with spastic equinus gait but with some ankle dorsiflexion movement, an articulated AFO with a plantar flexion stop can help improve the abnormal gait pattern. For children with a crouched gait, a ground reaction ankle–foot orthosis may help to decrease hyperdorsiflexion at the ankle, providing increased knee extension movement.
Upper limb orthoses may range from a custom molded resting hand splint to a neoprene sleeve at the wrist or hand to help prevent excessive wrist flexion and support the wrist in extension to facilitate grasp. Dynamic splints provide low-load continuous stretch to a joint and may be used to improve range of motion or to facilitate certain functional tasks.
Spinal orthoses vary from soft to rigid. The orthosis can provide compression and sensory feedback to facilitate trunk support in children with weak truncal musculature. For those with neuromuscular scoliosis, a properly fitting, supportive, custom seating device is favored over rigid spinal bracing, as the latter has not been proven to be effective in correcting the spinal curvature. Compliance is an issue as the brace requires at least 23 hours per day of wear.)
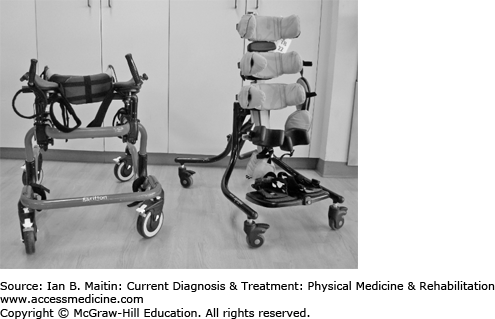
Options include custom seating devices, such as a medical stroller or wheelchair (manual or power), ramps, standers, gait trainers and walkers, and crutches (Figure 20–5). Lift systems are offered to families of children who are fully dependent in transfers when the child’s increasing weight makes such transfers difficult. Bath equipment (eg, shower chair, tub bench, and commode) also makes it safer and easier for children to perform or caregivers assist with activities of daily living. Home modifications may be needed to accommodate adaptive equipment, including installation of grab bars that will allow some independence in activities of daily living for household ambulators.
Spasticity management requires a team approach, which includes the patient (if able), caregiver, and rehabilitation team. Goals may include alleviation of pain, increase in function and mobility, improvement in range and flexibility, and assistance in alleviating caregiver burden. Providers should remember that underlying the abnormal muscle tone is weakness. Several treatment options can be used, singly or in combination, depending on the location, distribution, and severity of spasticity, and how it affects the patient’s function and care.
Oral medications are indicated for patients requiring generalized spasticity management. There is evidence supporting short-term spasticity treatment with diazepam in children with cerebral palsy, and some evidence supporting tizanidine. However, evidence is insufficient to support or refute improved motor function using these medications, or dantrolene or baclofen (oral or intrathecal). Table 20–4 lists common oral medications used for spasticity and dystonia management.
| Medication | Class | Dosage | Formulationa | Side Effects | Precautions |
|---|---|---|---|---|---|
| Baclofen | GABAB agonist | Initial: 10–20 mg/day divided 3 times daily; slowly titrate up Children 1–4 y: 2.5–5 mg 2–3 times daily Children 5–12 y: 2.5–10 mg 3 times daily Maximum: 80–120 mg/day | Tablet: 10 mg, 20 mg Oral suspension: 10 mg/mL (compounded) | Drowsiness, CNS depression, fatigue, weakness, constipation, decreased seizure threshold | Do not stop abruptly Hallucination, seizure, pruritus with abrupt withdrawal (especially with intrathecal delivery) |
| Diazepam | GABAA agonist | 0.12–0.8 mg/kg/day in divided doses every 6–8 h | Solution: 1 mg/mL Tablet: 2 mg, 5 mg | Drowsiness, CNS and respiratory depression, hypotension, fatigue, weakness Paradoxical reactions: hyperactive or aggressive behavior, hallucinations | Monitor respiratory status Abrupt withdrawal may cause increased seizure activity |
| Tizanidine | α2-Adrenergic agonist | Initial: 0.5–1 mg 3 times daily; slowly titrate up Maximum: 6 mg/day | Tablet: 2 mg, 4 mg | Drowsiness, fatigue, loss of appetite, nausea or vomiting (or both), nervousness, hallucinations, hypotension | Monitor respiratory status and blood pressure |
| Dantrolene sodium | Inhibits calcium release at sarcoplasmic reticulum of skeletal muscles | Initial: 0.5 mg/kg/dose 2 times daily; slowly increase Maximum: 3 mg/kg 4 times daily or 100 mg 4 times daily | Tablet: 25 mg, 50 mg, 100 mg | Lightheadedness, vertigo, weakness, malaise, diarrhea Potential hepatotoxicity | Monitor LFTs closely |
| Trihexyphenidylb | Anticholinergic | Initial: 0.05 mg/kg 2 times daily; increase gradually in increments of 0.05 mg/kg/day weekly up to 0.25.mg/kg 3 times daily Or Start with 1–2 mg/day; slowly titrate up to 15 mg/day | Tablet: 2 mg, 5 mg Syrup: 2 mg/5 mL | Drowsiness, dizziness, constipation, urinary retention, flushing, nausea, nervousness, blurred vision, dry mouth | Gradually increase dosage; monitor for anticholinergic side effects |
| Clonazepamb | Benzodiazepine (GABA agonist) | Initial: 0.01–0.03 mg/kg/day divided 2 or 3 times daily Maintenance: 0.1–0.2 mg/kg/day divided 2 or 3 times daily | Tablet: 0.5 mg, 1 mg, 2 mg Wafer: orally disintegrating tablets 0.125 mg, 0.25 mg, 0.5 mg, 1 mg, 2 mg | Drowsiness, dizziness, ataxia, fatigue, CNS depression | Do not stop abruptly Monitor LFTs and CBC if long-term use |
| Carbidopa/Levodopab | Dopamine agonist/precursor | 25/100 formulation: may start with ¼ tablet 2 times daily and gradually titrate up to 1 tablet 3 or 4 times daily | Tablet: 10/100 mg, 25/100 mg, 25/250 mg Can be compounded for elixir | Gastrointestinal upset, nausea, vomiting (significant limiting factor), sedation, dyskinesia | May give additional carbidopa, or give with meals to Minimize gastrointestinal symptoms |
In children, the recommended dose of botulinum toxin A used for chemodenervation is 16–20 units/kg, which may be repeated every 12 weeks, if needed. Dosing for different muscles to be injected depends on muscle size, severity of spasticity, and goals.
Alcohol blocks such as phenol, 3–6%, are also used for motor point or perineural injections. These are especially helpful when administered for obturator motor branch block to decrease hip adductor spasticity, or, in the musculocutaneous nerve, for upper limb spasticity. Effects are immediate and last longer than botulinum toxin. Side effects include pain over the injection site, dysesthesia, and possible tissue edema and scarring.
General indications for orthopedic surgery include fixed contractures and rotational abnormalities affecting function, joint dislocation with pain, and shoe wear and hygiene problems. Interventions include muscle–tendon lengthening, tendon transfers, osteotomies, and arthrodesis procedures. Muscle–tendon lengthening procedures may include Z-type tendon lengthening or muscle lengthening. Z-type tendon lengthening increases joint range of motion, whereas muscle lengthening results in the muscle recessing closer to the muscle tendon junction, which disrupts Golgi receptors and thus decreases spasticity. The side effects of Z-type tendon lengthening may include tendon overlengthening or a shortened muscle.
Tendon transfers utilize spastic muscles in order to fix a deformity by reattaching a spastic muscle to a weaker antagonist muscle group. A spastic rectus femoris may be transferred to the hamstrings to correct a stiff knee gait during swing phase. Or, a spastic anterior or posterior tibialis may be transferred to the lateral foot to help correct a varus foot deformity. Because tendon surgeries may need to be repeated, Graham and colleagues recommend waiting until a patient reaches 7–9 years of age before surgically intervening; this is a sufficient time for dystonia to completely appear, and also allows for a comprehensive gait analysis to be performed. A single-event multilevel surgery (SEMLS) may be performed that combines bone surgeries with tendon surgeries (which would otherwise likely need to be repeated if done prior).
An osteotomy involves cutting bone to improve position and alignment. For the child who has a dysplastic hip with subluxation and coxa valga, a varus derotation osteotomy to the proximal aspect of an anteverted femur can improve hip coverage, and superior placement of muscle and tendons will result in more power and improved energy conservation. An arthrodesis, or joint fusion, may be performed to correct an alignment deformity, stabilize a joint, or prevent recurrence of contracture.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


