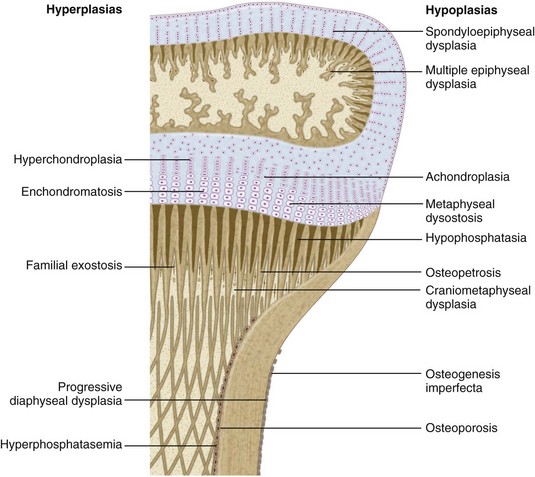
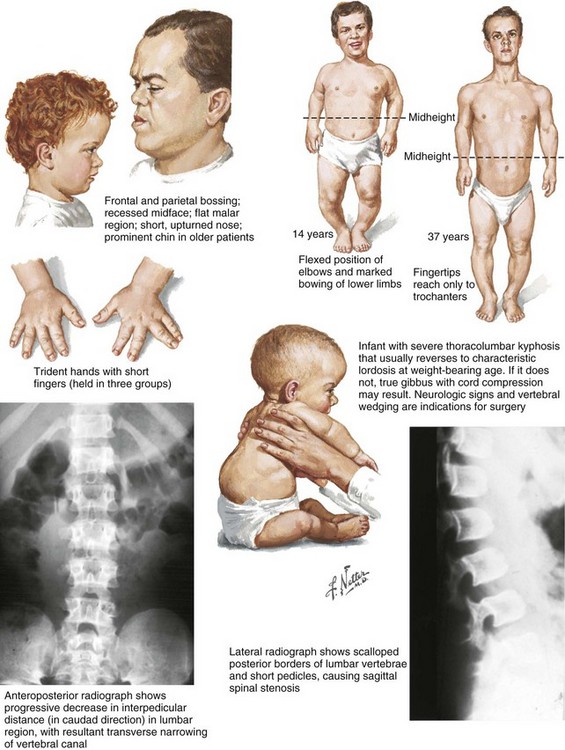
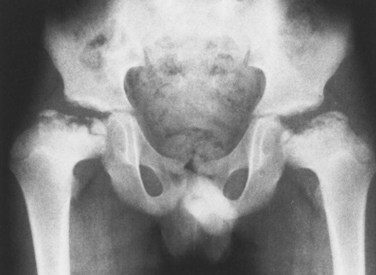
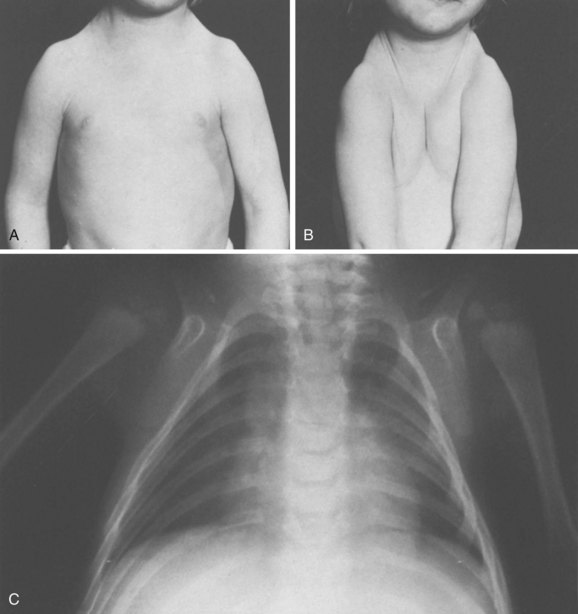
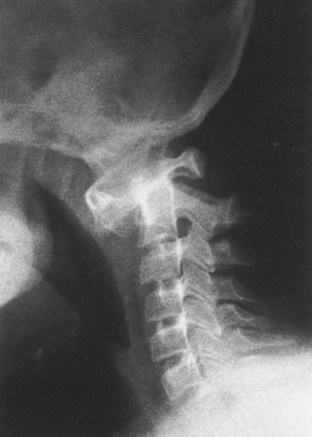
Chapter 3 SECTION 1 BONE DYSPLASIAS (DWARFISM) III. Spondyloepiphyseal Dysplasia VI. Metaphyseal Chondrodysplasia VII. Multiple Epiphyseal Dysplasia VIII. Dysplasia Epiphysealis Hemimelica (Trevor Disease) IX. Progressive Diaphyseal Dysplasia (Camurati-Engelmann Disease) XII. Cleidocranial Dysplasia (Dysostosis) SECTION 2 CHROMOSOMAL AND TERATOLOGIC DISORDERS SECTION 3 HEMATOPOIETIC DISORDERS SECTION 4 METABOLIC DISEASE/ARTHRITIDES III. Idiopathic Juvenile Osteoporosis V. Infantile Cortical Hyperostosis (Caffey Disease) IX. Juvenile Idiopathic Arthritis SECTION 7 NEUROMUSCULAR DISORDERS II. Myelodysplasia (Spina Bifida) III. Myopathies (Muscular Dystrophies) IV. Polymyositis and Dermatomyositis VII. Anterior Horn Cell Disorders VIII. Acute Idiopathic Postinfectious Polyneuropathy (Guillain-Barré Syndrome) SECTION 8 CONGENITAL DISORDERS II. Adolescent Idiopathic Scoliosis III. Infantile Idiopathic Scoliosis IV. Juvenile Idiopathic Scoliosis SECTION 10 UPPER EXTREMITY PROBLEMS SECTION 11 LOWER EXTREMITY PROBLEMS: GENERAL I. Developmental Dysplasia of the Hip III. Legg-Calvé-Perthes Disease (Coxa Plana) IV. Slipped Capital Femoral Epiphysis V. Proximal Femoral Focal Deficiency A Definition: Dysplasia means abnormal development. 1. Shortening of the involved bones affects specific portions of the growing bone (Figure 3-1); hence the term dwarfism. Most forms of dwarfism are related to gene defects (single or multiple genes; Table 3-1). Table 3-1 Pediatric Congenital Disorders and Associated Genetic Defects B The term proportionate dwarfism implies a symmetric decrease in both trunk and limb length (e.g., as occurs with mucopolysaccharidoses). 1. Short-trunk variety (e.g., Kniest syndrome–spondyloepiphyseal) 2. Short-limb variety (e.g., achondroplasia, diastrophic dysplasia) 1. Achondroplasia is the most common form of disproportionate dwarfism. 2. Autosomal dominant condition; 80% of cases caused by a spontaneous mutation in the fibroblast growth factor receptor 3 (FGFR3) 3. This disproportionate, short-limbed form of dwarfism is caused by abnormal endochondral bone formation that is more affected than appositional growth. 4. Anatomically, achondroplasia is categorized as a physeal dysplasia. 5. Caused by failure in the cartilaginous proliferative zone of the physis. Achondroplasia is a quantitative, not a qualitative, cartilage defect. 1. Normal trunk and short limbs (rhizomelic) with hypotonia 2. Frontal bossing, button noses, small nasal bridges, trident hands (inability to approximate extended middle and ring fingers) (Figure 3-2). 3. Thoracolumbar kyphosis (which usually resolves around the age at ambulation) 4. Lumbar stenosis (most likely to cause disability) and excessive lordosis (short pedicles with decreased interpedicular distances) 6. Normal intelligence but delayed motor milestones 7. Although sitting height may be normal, standing height is below the third percentile. 1. Lumbar stenosis: decompression and fusion of the spine for a developing neurologic deficit (usually in children older than age 10) 2. Foramen magnum stenosis: decompression 3. Progressive kyphosis: if fail bracing, anterior and posterior fusion are indicated for residual kyphosis greater than 60 degrees by age 5 years. 4. Genu varum: Tibial osteotomies or hemiepiphysiodesis 5. Limb lengthening through callodiastasis (lengthening through a metaphyseal corticotomy) have been well described, with a high rate of complications. D Pseudoachondroplasia: This disorder is clinically similar to achondroplasia. III SPONDYLOEPIPHYSEAL DYSPLASIA 1. Must be differentiated from multiple epiphyseal dysplasia (MED) 2. MED and spondyloepiphyseal dysplasia involve abnormal epiphyseal development in the upper and lower extremities. 3. The distinguishing feature of spondyloepiphyseal dysplasia is the added typical spine involvement. 1. Autosomal dominant; short-trunked, disproportionate dwarfism with joint stiffness/contractures, scoliosis, kyphosis, dumbbell-shaped femora, and hypoplastic pelvis and spine VI METAPHYSEAL CHONDRODYSPLASIA 1. Heterogeneous group of disorders characterized by metaphyseal changes of tubular bones with normal epiphyses 1. Jansen (rare): most severe form VII MULTIPLE EPIPHYSEAL DYSPLASIA 1. Short-limbed, disproportionate dwarfism that often is not manifested until between the ages of 5 and 14. It must be differentiated from spondyloepiphyseal dysplasia. A mild form (Ribbing) and a more severe form (Fairbanks) exist. 1. MED is characterized by irregular, delayed ossification at multiple epiphyses (Figure 3-3). 2. Short, stunted metacarpals and metatarsals, irregular proximal femora, abnormal ossification (tibial “slant sign” and flattened femoral condyles, patella with double layer), valgus knees (early osteotomy should be considered), waddling gait, and early hip arthritis are common. 3. The proximal femoral involvement can be confused with Perthes disease. VIII DYSPLASIA EPIPHYSEALIS HEMIMELICA (TREVOR DISEASE) IX PROGRESSIVE DIAPHYSEAL DYSPLASIA (CAMURATI-ENGELMANN DISEASE) 1. Autosomal dominant inheritance. 2. Affected children are often “late walkers” (because of associated muscle weakness), with symmetric cortical thickening of long bones. 3. The tibia, femur, and humerus are most often involved (in that order), affecting only the diaphyseal portion of bone. 1. In contrast to the aforementioned conditions, these forms of dwarfism are easily differentiated on the basis of the presence of complex sugars in the urine. 2. The accumulation of mucopolysaccharides, as a result of a hydrolase enzyme deficiency, produces a proportionate dwarfism. 1. Morquio syndrome (autosomal recessive) 2. Hurler syndrome (autosomal recessive inheritance) 3. Hunter syndrome (sex-linked recessive inheritance) 4. Sanfilippo syndrome (autosomal recessive inheritance) 1. Autosomal recessive inheritance; severe, short-limbed dwarfism 2. Cleft palate (59% of cases) 3. Severe joint contractures (especially hip and knee) 4. Cauliflower ears (80% of cases), hitchhiker’s thumb 1. Spine radiographs reveal cervical kyphosis (often severe, necessitating immediate treatment), thoracolumbar kyphoscoliosis (83% of cases), spina bifida occulta, and atlantoaxial instability. XII CLEIDOCRANIAL DYSPLASIA (DYSOSTOSIS) 1. Autosomal dominant inheritance 2. Proportionate dwarfism that affects bones formed by intramembranous ossification (clavicles, cranium, pelvis) 1. Hypoplasia or aplasia of the clavicle (no intervention necessary) (Figure 3-4) 2. Delayed closure of skull sutures 5. Delayed ossification of the pubis XIII DYSPLASIAS ASSOCIATED WITH BENIGN BONE GROWTH 1. Usually characterized by ligamentous laxity, hypotonia, mental retardation, heart disease with atrial septal defect (50% of cases), endocrine disorders (hypothyroidism and diabetes), and premature aging 2. Orthopaedic problems include metatarsus primus varus, pes planus, spinal abnormalities (atlantoaxial instability [Figure 3-5], scoliosis [50% of cases], spondylolisthesis [6% of cases]), hip instability (open reduction with or without osteotomy is usually required), slipped capital femoral epiphysis (SCFE) (hypothyroidism should be sought), patellar dislocation, and symptomatic planovalgus feet. 3. Atlantoaxial instability may be subtle but commonly manifests as a loss or change in motor milestones. 1. Trisomy 21 is the most common chromosomal abnormality; its incidence increases with maternal age. 2. Chromosome 21 is the location of genes that encode for type VI collagen (COL6A1 and COL6A2). 1. Lower extremity radiographs are needed to evaluate for patella dislocations and genu valgum. 2. Anteroposterior and frog-leg pelvic views to evaluate for SCFE 3. Flexion-extension radiographs of the cervical spine to evaluate atlantoaxial instability 1. Screening for cervical spine instability: 2. Children with asymptomatic instability should avoid contact sports, diving, and gymnastics. 3. Children with symptomatic instability often require surgery, but the rate of wound healing problems and infection is high. 1. Affected patients are female and have short stature, lack of sexual development, webbed neck, and cubitus valgus. 2. Idiopathic scoliosis is common. Growth hormone therapy can exacerbate scoliosis. 3. Malignant hyperthermia is common with anesthetic use. 4. Must be differentiated from Noonan syndrome (same appearance except for normal gonadal development, mental retardation, and more severe scoliosis) 1. Genu valgum and shortening of the fourth and fifth metacarpals, which usually necessitate no treatment 1. Floppy, hypotonic infant who grows up to be an intellectually impaired, obese adult with an insatiable appetite 1. Characteristic “kinky” hair 2. May be differentiated from occipital horn syndrome (which also affects copper transport) in that the latter is characterized by bony projections from the occiput of the skull 1. Progressive impairment and stereotaxic, abnormal hand movements (like those in autism) 2. Manifests in girls at 6 to 18 months of age 3. Loss of developmental milestones that is rapid and then stabilizes 1. Family of deletion mutations of the X-linked gene encoding a protein called methyl-CpG-binding protein 2 (MECP2) VI BECKWITH-WIEDEMANN SYNDROME 1. Organomegaly, omphalocele, and a large tongue 2. Orthopaedic manifestations include hemihypertrophy with spastic cerebral palsy. 3. There is a predisposition to Wilms tumor (patient must be screened regularly with kidney ultrasonography). 1. Spasticity is thought to be the result of infantile hypoglycemic episodes secondary to pancreatic islet cell hypertrophy. VII TERATOGEN-INDUCED DISORDERS A Fetal alcohol syndrome: Maternal alcoholism can cause growth disturbances, central nervous system dysfunction, dysmorphic facies, hip dislocation, cervical spine vertebral and upper extremity congenital fusions, congenital scoliosis, and myelodysplasia. Contractures respond to physical therapy. B Maternal diabetes: This may lead to heart defects, sacral agenesis, and anencephaly. Careful management of pregnant diabetic women is essential. C Other teratogens: These include drugs (e.g., aminopterin, phenytoin, thalidomide), trace metals, maternal conditions, infections, and intrauterine factors; they may also lead to orthopaedic manifestations in affected children. 2. Bone pain (Gaucher crisis), and bleeding abnormalities 3. Hepatosplenomegaly (characteristic finding) 1. Aberrant autosomal recessive, lysosomal storage disease characterized by accumulation of cerebroside in cells of the reticuloendothelial system. The cause is a deficiency of the enzyme β-glucocerebrosidase. 1. Metaphyseal enlargement (failure of remodeling), femoral head necrosis (may be confused with Perthes disease or MED), “moth-eaten” trabeculae, patchy sclerosis, and Erlenmeyer flask deformity of the distal femora (70% of cases)
Pediatric Orthopaedics
section 1 Bone Dysplasias (Dwarfism)

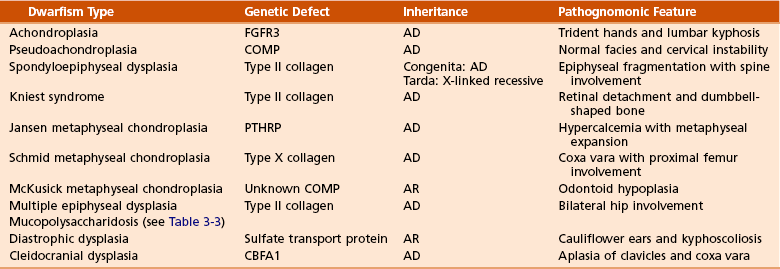
Disorder
Genetic Defect
Achondroplasia
FGFR3
Hypochondroplasia
FGFR3
Thanatophoric dysplasia
FGFR3
Pseudoachondroplasia
COMP
Multiple epiphyseal dysplasia type I
COMP
Multiple epiphyseal dysplasia type II
Collagen type IX
Spondyloepiphyseal dysplasia congenita
Collagen type II
Kniest syndrome
Collagen type II
Stickler syndrome (hereditary arthro-ophthalmopathy)
Collagen type II
Diastrophic dysplasia
Sulfate transporter gene
Schmid metaphyseal chondrodysplasia
Collagen type X
Jansen metaphyseal chondrodysplasia
PTHRP
Craniosynostosis
FGFR2
Cleidocranial dysplasia
CBFA1
Hypophosphatemic rickets
PEX
Marfan syndrome
Fibrillin-1
Osteogenesis imperfecta
Collagen type I
Ehlers-Danlos syndrome
Types I and II
Collagen type V
Type IV
Collagen type IV
Types VI and VII
Collagen type I
Duchenne/Becker muscular dystrophies
Dystrophin
Limb-girdle dystrophies
Sarcoglycan and dystroglycan complex
Charcot-Marie-Tooth disease
PMP22
Spinal muscular atrophy
Survival motor neuron protein
Myotonic dystrophy
Myotonin
Friedreich ataxia
Frataxin
Neurofibromatosis
Neurofibromin
McCune-Albright syndrome
cAMP
 Neurologic symptoms are usually related to nerve root or spinal cord compression, which can occur at any level, including the foramen magnum (which may cause periods of apnea).
Neurologic symptoms are usually related to nerve root or spinal cord compression, which can occur at any level, including the foramen magnum (which may cause periods of apnea).
 Genetic defect is in parathyroid hormone–related peptide.
Genetic defect is in parathyroid hormone–related peptide.
 Striking bulbous metaphyseal expansion of long bones is a distinctive radiographic finding.
Striking bulbous metaphyseal expansion of long bones is a distinctive radiographic finding.
 Most common form; manifests by ages 18 months to 2 years with waddling gait, genu valgum (“knock-knees”), thoracic kyphosis, cloudy corneas, and normal intelligence
Most common form; manifests by ages 18 months to 2 years with waddling gait, genu valgum (“knock-knees”), thoracic kyphosis, cloudy corneas, and normal intelligence
 Urinary excretion of keratan sulfate
Urinary excretion of keratan sulfate
 Urinary excretion of dermatan/heparan sulfate
Urinary excretion of dermatan/heparan sulfate
 Bone marrow transplantation has increased the life span for patients with this disorder.
Bone marrow transplantation has increased the life span for patients with this disorder.
 Urinary excretion of dermatan/heparan sulfate
Urinary excretion of dermatan/heparan sulfate
 Bone marrow transplantation has increased the life span for patients with this disorder.
Bone marrow transplantation has increased the life span for patients with this disorder.
section 2 Chromosomal and Teratologic Disorders
section 3 Hematopoietic Disorders
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pediatric Orthopaedics