Chapter 5 Pain-processing mechanisms in whiplash associated disorders
Introduction
It is now generally acknowledged that an initial peripheral injury of some kind occurs to the neck following whiplash injury,1 although the specific injured structure in individual patients is difficult to clinically identify with current imaging techniques.2 Regardless of the specific structures involved, following injury and ensuing inflammation, a cascade of events occurs in the periphery, spinal cord and supraspinal nervous centres leading to upregulation of nociceptive processes. These changes have the potential to amplify the individual’s pain experience.
Even if identification of injured peripheral (neck) structures was easily achieved, it remains unclear whether it would improve management directions and outcomes, as the correlation between objective findings and pain and disability is well known to be equivocal in patients with whiplash.3, 4 Rather, recent calls have been made for greater consideration of underlying pain processes that contribute to the condition in question, as this may enable the direction of the most appropriate intervention strategies.5 Research has now demonstrated indirect evidence, via sensory testing and other methods, of altered pain processes as contributing factors to the clinical presentation, outcome and response to treatment of whiplash associated disorders (WAD).
Mechanisms for post-injury central hyperexcitability
Peripheral mechanisms
Tissue injury leads to an inflammatory response, with the release of different mediators, such as potassium ions, substance P, bradykinin and prostaglandins.6 These substances may induce a sensitisation of peripheral receptors, with changes in the response characteristics of primary afferent fibres.7 They may also activate normally inactive or ‘silent’ nociceptors.8 Furthermore, the inflammatory response induces a gene expression in the dorsal root ganglion, resulting in an increased synthesis of peripheral receptors, which contributes to the increased sensitivity of the nociceptor.9 Long-lasting nociceptive stimulation may lead to a modification in the peripheral fibres: Aβ-fibres may start synthesising receptors that are normally found only in C-fibres, with the Aβ-fibre adopting C-fibre characteristics.10
These sensitising events mediate primary hyperalgesia,11 in which a reduced threshold for eliciting pain and enhanced pain to supra-threshold stimuli within the injured area can be recorded. Peripheral sensitisation ultimately results in an increased nociceptive input to the spinal cord.
Spinal cord plasticity
Afferent nociceptive input induces an increase in the excitability of central sensory neurons,12 which is mediated by the N-methyl-D-aspartate (NMDA) receptor.13, 14 The expression of the isoenzyme cyclooxygenase-2 (COX-2) in the spinal cord is also involved in spinal cord hyperexcitability.15 Importantly, COX-2 expression is not confined to the neural structures connected to the site of inflammation, but involves the whole spinal cord and the supraspinal centres.16 This phenomenon may be responsible, at least in part, for a generalised hypersensitivity to peripheral stimulation, such as the one evoked after stimulation of tissues that are a distance from the site of injury.
In animal models using direct neuronal measurements, peripheral tissue or nerve damage induced an expansion of receptive fields on individual dorsal horn neurons.17 This phenomenon represents an important manifestation of central sensitisation, leading to increased nociceptive input into the CNS and expansion of areas of referred pain and hyperalgesia.
Inflammation and nerve injury also produce depression of the inhibitory mechanisms within the spinal cord, with consequent amplification of the nociceptive signal being transmitted to higher centres.18 An important substrate of such disinhibition is the loss of inhibitory control of GABAergic (gamma-aminobutyric acid) and glycinergic spinal cord neurons that follows inflammation and nerve injury.19 Furthermore, nerve damage leads to destruction of inhibitory interneurons, which contributes to hyperexcitability.20
Endogenous modulation
Spinal cord hyperexcitability elicited by trauma or inflammation is influenced by descending facilitatory and inhibitory pathways.21 Opioidergic,22 serotonergic23 and noradrenergic systems23 are involved in descending inhibition.
Pathways that excite 5-hydroxytryptamine-3 (5-HT-3) receptors in the whole spinal cord after regional peripheral damage are involved in descending facilitation.24 Their activity could be driven by anxiety and fear, which are common features in patients with pain.25 These data provide an additional explanation for the widespread pain hypersensitivity following a peripheral lesion, a common phenomenon in patients with chronic pain.26 They may also represent a physiological basis for an amplification of the pain experience in patients with psychological distress.
The efficiency of the endogenous modulatory system can be tested in humans by diffuse noxious inhibitory control (DNIC), whereby a conditioning painful stimulus is expected to attenuate the response to an additional test stimulus. DNIC has been found to be altered in different pain syndromes, such as fibromyalgia,27 osteoarthritis of the hip28 and irritable bowel syndrome.29 Alteration in DNIC before surgery has been found to be a predictor of chronic post-thoracotomy pain.30
Brain plasticity
Several clinical studies have revealed that the brain is capable of substantial plastic changes. Using equal levels of sensory stimulation in patients with chronic low back pain and pain-free controls, patients with chronic low back pain showed not only more pain than healthy controls, but also more extensive patterns of neuronal activation in pain-related cortical areas.31 This confirmed the occurrence of enhanced central pain processing.
Patients with various chronic pain syndromes, such as phantom limb pain, low back pain and complex regional pain syndrome (CRPS), display substantial cortical reorganisation, whereby the representation of the different body areas is altered.32–34
Using magnetic resonance imaging (MRI), patients with chronic low back pain were found to have 5–11% less neocortical grey matter volume than control subjects.35 The decreased volume was related to the pain duration, indicating a 1.3 cm3 loss of grey matter for every year of chronic pain. These concerning findings indicate that chronic low back pain is accompanied by brain atrophy, although the clinical implications remain unclear.
Evidence for central hyperexcitability in chronic WAD
In a first investigation comparing whiplash patients with healthy controls, patients displayed lower pain detection and tolerance thresholds after cutaneous electrical stimulation of the neck.36 The absence of tissue damage at the site of testing suggested that a central sensitisation of nociceptive pathways was the cause of the pain hypersensitivity.
This finding has been confirmed by a study testing muscle pain sensibility after injection of hypertonic saline into the supraspinatus and tibialis anterior muscle.37 Patients with whiplash displayed higher pain scores, longer duration of pain and larger areas of local and referred pain after intramuscular injection of hypertonic saline, compared with healthy controls. In particular, the areas of referred pain were impressively larger in patients than in controls, at both the neck and leg. Several patients reported pain spreading to the whole leg and on the contralateral side, which was not the case in healthy subjects. These data suggest that pain hypersensitivity is not limited to the injured and surrounding areas (primary and secondary hyperalgesia), but may be generalised to the whole CNS. The impressive spread of referred pain is strongly suggestive of central hyperexcitability, possibly involving disinhibitory processes and expansion of receptive fields.
In a following investigation, pain thresholds to electrical and heat stimulation, applied at the skin and muscles of both the neck and a lower limb, were measured.26 Patients displayed lower pain thresholds with both cutaneous and muscular electrical stimulation, applied at both the neck and the lower limb, compared with a group of healthy subjects. This confirmed the previous findings of generalised central hypersensitivity. Infiltration of a local anaesthetic into the painful and tender muscles did not reduce either neck pain or pain thresholds. This indicates that the source of pain was not located in the infiltrated muscles and central hypersensitivity was not maintained by a nociceptive input arising from these muscles.
A study that analysed responses to a variety of non-painful stimuli in whiplash patients and healthy controls found that 65% of patients, but no control subject, reported pain after vibration stimulus.38 Patients reacted with more pain to heat and cold stimulation than healthy subjects. Because sensory stimuli were applied to non-damaged tissues, the results of this study provided additional evidence for the presence of central hyperexcitability in whiplash patients.
In a study that analysed spinal hyperexcitability using the nociceptive withdrawal reflex, patients with whiplash injury and patients with fibromyalgia displayed lower reflex thresholds than healthy subjects.39 These findings provide objective electrophysiological evidence for generalised spinal cord hyperexcitability.
A recent study addressed the question of the relationship between parameters of central hypersensitivity and psychological factors.40 While there was a weak correlation between catastrophising and cold pain thresholds, spinal excitability as assessed by the nociceptive withdrawal reflex did not correlate significantly with either catastrophising or psychological distress. A more recent investigation on 300 healthy volunteers found no impact of depression, anxiety and catastrophising on the nociceptive withdrawal reflex, indicating that spinal cord excitability is little or not affected by descending psychological influences.41
It is important to recognise that central hypersensitivity is not specific for patients with whiplash, but has been observed in different chronic pain syndromes, such as endometriosis,42 fibromyalgia,43 osteoarthritis,44 tension-type headache,45 temporomandibular joint pain46 and post-mastectomy pain.47
Evidence for central hyperexcitability in acute WAD and in the transition to chronicity
Most investigations of the nociceptive processes involved in WAD have focused on the chronic stage of the condition. However, recent data demonstrate that sensory disturbances observed in patients with chronic WAD are, in fact, present from soon after injury. In the acute phase post injury, local cervical mechanical hyperalgesia (decreased pressure pain thresholds) is found in individuals with both lower and higher levels of pain and disability.48, 49 This local hyperalgesia tends to resolve within a few months in individuals with good and fair recovery but persists unchanged in those who report ongoing moderate-to-severe symptom levels six months to two to three years post accident.50 Mechanical hyperalgesia is also found at sites away from the site of injury, including the upper and lower limbs.48, 49 The absence of tissue damage at the site of testing suggests the involvement of central sensitisation of nociceptive pathways. In contrast to local hyperalgesia, which occurs irrespective of pain and disability levels, the presence of generalised hyperalgesia has been shown to be more apparent in those individuals reporting higher pain and disability,49 and subsequent poor recovery.50 In addition to mechanical hyperalgesia, both cold and heat hyperalgesia have also been found to be features of individuals with higher symptom levels and poor functional recovery (Fig 5.1).50
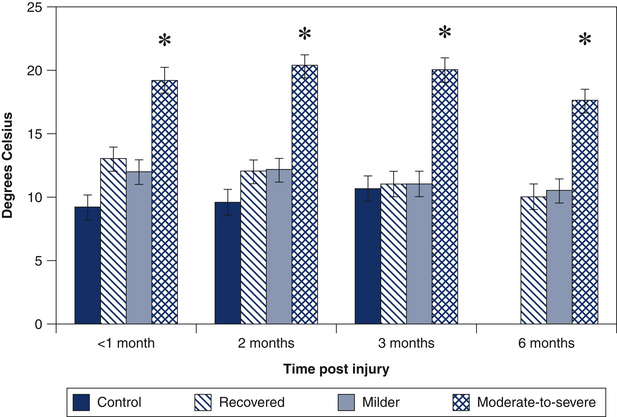
Figure 5.1 Cold pain thresholds (measured over the cervical spine) in controls (1–3 months) and three whiplash groups classified at six months post injury (recovered: nerve disability index [NDI] <8; milder symptoms: NDI: 10–28; and moderate-to-severe symptoms: NDI: >30) at 1, 2, 3 and 6 months post injury.50 The whiplash group with persistent moderate-to-severe symptoms demonstrates cold hyperalgesia from within a month of injury that does not resolve. ∗ = P < 0.05.
(Adapted from Sterling et al.50)
Most studies have explored the presence of positive sensory signs or sensory hypersensitivity. Recently, the presence of negative sensory responses or widespread hypoaesthesia (or elevated detection thresholds) occurring concurrently with hypersensitivity has also been found in patients with acute WAD.51 Similar to findings of sensory hypersensitivity, the hypoaesthetic changes were widespread and occurred in response to a variety of stimuli, including vibration (Fig 5.2), and thermal and electrical stimulation.51 While they occurred in the majority of participants in the acute injury stage (three to four weeks), the hypoaesthesia persisted only in those who were initially classified as at high risk of poor recovery (higher pain and disability and sensory hypersensitivity).52 Consideration of these findings of both positive and negative sensory changes to various stimuli, which are widespread and generalised throughout various body locations, infers that disturbances in both excitatory and inhibitory CNS processes are at play.
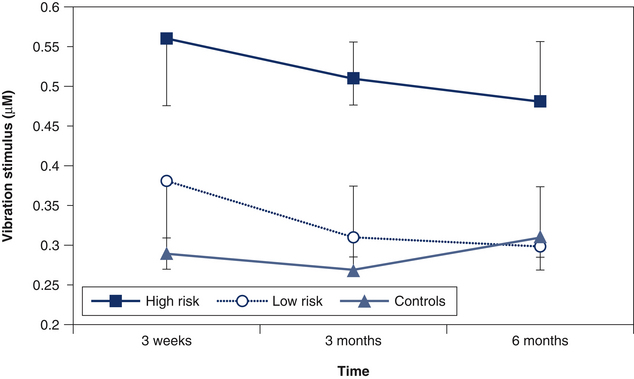
Figure 5.2 Vibration thresholds (mean and standard deviation) in the control, low- and high-risk whiplash groups over time. The stimulus was applied over areas of the hand innervated by C6–C7 (palmar 2nd metacarpal) dermatomes.52 (P < 0.05)
Importantly, some of the sensory phenomena demonstrate an ability to predict individuals at risk of poor recovery. In particular, the early presence of cold hyperalgesia is emerging as a consistent prognostic factor. Initial studies demonstrated that in addition to initial moderate pain, decreased neck movement, older age and post-traumatic stress symptoms, cold hyperalgesia predicted higher levels of pain and disability at both six months and two to three years post injury.53, 54 Cold pain tolerance measured using the cold pressor test has also shown predictive capacity.55 More recently, it has been shown that when injured people are classified based on the presence of moderate or greater initial pain and sensory hypersensitivity, 86% of those deemed at high risk of poor recovery did indeed develop persistent symptoms at six months post injury.52 Further research using trajectory modelling has validated cold hyperalgesia as a predictor of a clinical pathway to chronicity.56
While sensory hypersensitivity is thought to reflect augmented central pain processes, the precise underlying mechanisms are not clear. A recent prospective study investigating the nociceptive withdrawal reflex in patients progressing from acute to chronic WAD showed a different picture to what is seen for sensory hypersensitivity.57 Lowered reflex thresholds indicative of spinal cord hyperexcitability were found to be present at three weeks post injury in all whiplash subgroups, irrespective of the extent of recovery. However, by six months post injury, lowered reflex thresholds persisted only in those with moderate-to-severe symptoms, having resolved in those who recovered or reported lesser symptoms at this point in time. In contrast, generalised sensory hypersensitivity (pressure and cold) was only ever present in those with persistent moderate-to-severe symptoms and remained unchanged throughout the study period. This suggests different mechanisms underlie sensory hypersensitivity and nociceptive flexion responses (NFRs), or at least that sensory hypersensitivity does not appear to be solely due to spinal processes.
In summary, current evidence would suggest that some CNS pain processes are augmented from soon after injury in those individuals who do not recover but develop chronic moderate-to-severe pain and disability. The reasons as to why this group manifests more profound changes in pain processes is not clear but there are numerous possibilities, including but not limited to: the nature, extent and duration of the original injury providing peripheral nociceptive input to the CNS;6 stress-related responses;58 psychological augmentation;59 poorer health prior to the injury;60 or a genetic predisposition.61 Irrespective of the cause of the changes, the data indicate that consideration of these processes in the early management of WAD will be required.
Related posts:
 Epidemiology of whiplash associated disorders
Epidemiology of whiplash associated disorders
 Potential role of stress systems in the pathogenesis of whiplash associated disorders
Malingering and symptom magnification in whiplash associated disorders
Future directions
Potential role of stress systems in the pathogenesis of whiplash associated disorders
Malingering and symptom magnification in whiplash associated disorders
Future directions
 Neuromuscular dysfunction in whiplash associated disorders
Neuromuscular dysfunction in whiplash associated disorders
 The evidence for pathoanatomical lesions
The evidence for pathoanatomical lesions
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


