Fig. 19.1
Schematic illustration of an axial view of cervical spine anatomy, including soft and hard tissues with the potential for generating pain. The articulating intervertebral disc and facet joints limit motion and transmit loads, but also have the capacity for pain, due to their innervation. Neural components, such as the spinal cord, nerve roots and dorsal root ganglion (DRG), are also at risk for painful injury. The insets provide immunohistochemical images showing the presence of nerve fibers (positive for CGRP) in the intervertebral disc (brown), dorsal root (red), and facet capsule (green)
Many of these structures have innate populations of nociceptive fibers [5–8], supporting their ability to generate pain when such fibers are activated during neck and/or tissue loading. Because of their potential for injury and highly likely contribution to pain generation, we briefly review them here, in that context. However, a detailed review of these anatomical structures can be found elsewhere [9, 10]. In addition, for the major tissue components we also provide a brief discussion of the potential loading scenarios that may initiate pain and also relate relevant anatomy to those injury conditions. In a later section we particularly focus on specific tissues and the relationship between their injury, pain and the nociceptive mechanisms that are affected by mechanical contributions.
19.2.1 Bone, Intervertebral Disc, and Ligaments
The cervical spine includes seven vertebrae (C1–C7), with C1 and C2 (the atlas and axis, respectively) retaining specialized function since they interface with the base of the skull and each other. Each pair of bony vertebrae of the cervical spine are coupled by the collagenous intervertebral discs anteriorly and the bilateral articulating facet joints posterolaterally (Fig. 19.1). The vertebrae themselves are innervated most extensively in the periosteum that lines the outer surface of each vertebra [5]. Large intraosseus nerves and neurovascular bundles permeate the vertebral body, penetrating to central and peripheral regions including the marrow and the cartilaginous endplates that are adjacent to the intervertebral disc [5, 11].
The intervertebral disc is a collagenous structure that connects to its adjacent vertebral bodies via a cartilaginous endplate (Fig. 19.1). The disc consists of an outer annulus fibrosus made up of concentric rings of stiff fibrocartilage and an inner, gelatinous nucleus pulposus [12]. The primary function of the intervertebral disc is to absorb and transmit axial compressive loading through the spinal column and to control loading by distributing stresses within the dense fibrocartilage of its annulus fibrosus. However, its neuroanatomy suggests that it may also have the capacity for pain generation, owing to the fact that the outer one-third of the annulus fibrosus of the normal asymptomatic disc is innervated by peptidergic fibers and free nerve terminals, which are unencapsulated nerve endings that often respond to noxious stimuli (Fig. 19.1 inset) [7, 11, 13, 14]. These nerve terminals can be activated by a variety of stimuli, including altered disc loading by injury and spinal degeneration, or even from exposure to chemical mediators [13, 15]. Despite evidence of disc innervation by sensory fibers, and a recent focus on the hypothesis of disc-mediated pain from aberrant nerve fiber in-growth in discogenic pain conditions [16–18], there are very limited studies defining the cellular mechanisms leading to persistent pain that arises from the disc after its undergoing abnormal loading. For example, whole body vibration in the rat that produces pain has been shown to also increase neurotrophins (i.e. NGF, BDNF) in the disc [19, 20]. Nonetheless, the effects of aging and degeneration of the disc and the associated anatomical changes in, and from, the vertebrae are a major pathway for initiating pain from this spinal tissue, particularly in the lumbar spine [14, 16, 21].
The cervical vertebrae are stabilized by many ligaments in both the anterior and posterior regions of the spine (Fig. 19.1) that normally contribute to maintaining the head position and providing neck proprioception, which is the sensory input that aids in awareness of relative position and movement of the body during spinal motions. The ligaments in the cervical spine include the anterior and posterior longitudinal ligaments that span the entire vertebral column, ligaments that connect pairs of adjacent vertebrae (ligamentum flavum, interspinous, and supraspinous ligaments), and the bilateral facet capsular ligaments that enclose the apposing surfaces of the articular pillars of adjacent vertebrae [22, 23]. The vertebrae of the upper cervical spine have additional ligaments that facilitate and restrict articulations between the atlas (C1) and axis (C2), as well as with the occiput [22]. The anterior longitudinal, facet capsular, supraspinous, and interspinous ligaments are at particular risk for undergoing excessive strain during injurious loading of the cervical spine, because of their role in bearing tensile loads during spinal motion [24, 25].
These ligaments have a diverse morphology according to their function, but even with their varied roles and structures, they are quite similar in their neuroanatomical features [8]. Within their dense collagenous networks, spinal ligaments are innervated by both proprioceptive and nociceptive bundles, including small diameter sensory fibers with free nerve endings that respond to noxious stimuli (Fig. 19.1 inset) [8, 26, 27]. Numerous immunohistochemical studies have reported the longitudinal, supraspinous, interspinous, and facet capsular ligaments to be innervated by nociceptive fibers and free nerve endings [27–32]. Histological studies have also identified low and high threshold mechanoreceptors, based on visualization of specialized sensory terminals, in many of these same spinal ligaments, along with nociceptive innervation [29, 31, 33–35]. The capacity of nerve fibers in spinal ligaments to be activated by a wide range of mechanical loading modalities further suggests their role in initiating and/or signaling painful loading from cervical spine injuries due to excessive loading of those tissues.
The facet capsular ligaments enclose the facet joints, which are bilateral articulations between each pair of adjacent vertebrae that contain a variety of innervated structures with the potential to generate pain (Fig. 19.1). The medial branches of the dorsal primary ramus innervate the cervical facet joints from the two levels superior and inferior to the joint [36], with mechanoreceptive and nociceptive fibers terminating in the joint and its capsule [6, 34, 37]. The presence of these afferents in the facet joints suggests that this joint has the potential to produce pain under abnormal and/or excessive loading scenarios. Furthermore, clinical studies have demonstrated the involvement of the facet joint in neck pain both by inducing pain through joint provocation (either by distention or injection of chemically irritating agents) and relieving pain through nerve ablation or anesthetic blockade [38–42]. The articulating surfaces of the joint themselves are covered with articular cartilage, and synovial folds exist in the joint space to dissipate stress and maintain stability. Synovial folds in cervical joints are believed to contribute to neck pain and headache following trauma [43, 44], and recent morphological studies have opined several hypotheses for injury mechanisms [45]. Taken together, the complicated biomechanics associated with many types of neck loading, the potential simultaneous activation of pain afferents in facet joint’s synovial folds and facet capsule, and the multifaceted clinical evidence all strongly suggest this joint as a primary anatomical source of neck pain due to its, or the spine’s, injury.
The bony and soft tissue structures in the cervical spine articulate and interact to support movement and distribute and absorb loading to the various individual tissues. Many biomechanical studies using in vivo and post-mortem human spinal tissues provide a detailed understanding of cervical spine biomechanics under many physiologic and injurious loading conditions [46–50]. During normal loading of the spine, the intervertebral disc and the complement of spinal ligaments guide and restrict motions of the head and neck within physiologic limits. The disc absorbs and distributes the compressive forces that are applied to the anterior column of the spine during flexion or extension. The spinal ligaments serve to resist tensile loading, while their proprioceptive and nociceptive innervation provide sensory feedback. The facet capsular ligament and its role in spinal loading, injury, and pain will be discussed in a later section.
The anatomical structures of the cervical spine can be injured or can fail completely when the compressive, tensile, or shear loading that is experienced by a single tissue exceeds its tolerance during extreme loading scenarios. For example, the disc is particularly susceptible to shear loading and torsion, especially without protection from the facet joints [50]. Likewise, the spinal ligaments have widely varying failure strengths [51, 52], but excessive spinal flexion or extension can exceed those limits, causing their rupture and activating nociceptive signaling pathways. These injuries trigger complex pain cascades and initiate a variety of different cellular mechanisms, both in the injured tissue and in the peripheral nervous system (PNS) and the CNS [53, 54]; relevant pain mechanisms are discussed in greater detail in the later Sect. 19.3.
19.2.2 Spinal Cord, Nerve Root, and Dorsal Root Ganglion (DRG)
The spinal cord is enclosed by a system of three elastic, collagenous membranes making up the meninges: the innermost pia mater, the arachnoid mater, and the outermost dura mater. The dura and arachnoid mater lie directly apposed to each other, separating the vertebral column from the spinal cord and the cerebrospinal fluid within. This dual membranous layer has a longitudinally-oriented collagenous structure that conveys substantial tensile strength along that axis, along with elastic fibers that maintain flexibility [12, 55]. At each spinal level, nerve roots terminate in the spinal cord within the meningeal layers. As they course peripherally, the nerve roots traverse the dura mater and exit the vertebrae, where a separate, albeit less mechanically-robust structure (the epineurium), envelops them [12]. The dura mater contains small, peptidergic nociceptive nerve fibers that express the neuropeptides calcitonin gene-related peptide (CGRP) and substance P [32, 56, 57]. Compared to the cranial meninges, the spinal dura mater has few of these fibers, and so, may play a limited role in the pathogenesis of pain [56]. However, the meninges may have a role in modulating pain through their ability to release pro-inflammatory cytokines into the cerebrospinal fluid that bathes the tissues of the CNS [58].
The spinal cord itself is highly organized and composed of white matter and gray matter (Fig. 19.1). White matter consists largely of myelinated axonal tracts conveying ascending and descending signals to and from the brain [12]. White matter tracts are found at the periphery of the spinal cord and somatotopically organized in the dorsal, ventral, and lateral columns. In contrast, the gray matter includes the neuronal cell bodies, unmyelinated axons, and dendritic structures. The gray matter is divided into the dorsal and ventral horns, with the commissural and intermediate gray matter connecting the horns centrally. The gray matter of the spinal cord is arranged in layers, or laminae, each containing distinct populations of neurons and receiving input from specific groups of fibers [59]. For example, laminae I and II along the superficial most dorsal boundary of the gray matter contain synaptic terminations of exclusively nociceptive fibers [60]. Afferent sensory fibers and descending motor axons enter and exit the spinal cord through the dorsal and ventral rootlets that originate in the gray matter [12].
The spinal cord is involved in the processing and amplification of painful sensory input from peripheral tissues. The innate potential of the spinal cord to generate pain is derived from damage to the nervous tissue itself, since it is comprised of neuronal processes and has no explicit sensory nerve endings. Spinal cord injuries (SCI) are diverse and have many potential causes due to mechanical injury, including partial or complete transection of the cord, contusion from blunt impact or pressure from broken bones or herniated discs, and sharp incision [61–65]. Each of these injuries can lead to varying degrees of cell death, demyelination, and axonal disruption and degeneration due to the initial mechanical insult, accompanied by secondary neuronal, immune and inflammatory responses [12, 66, 67]. Although motor deficits are often a primary outcome of spinal cord injury, widely varying pathogenic pain states often accompany motor deficiency and their cause and mechanisms remain an active area of research [68, 69].
Rootlets come off the spinal cord both anteriorly and posteriorly and combine to form the dorsal and ventral nerve roots that comprise the bilateral peripheral spinal nerves at each level (Fig. 19.1). In general, the posterior dorsal rootlets are afferent sensory fibers and the anterior ventral rootlets are effecter motor fibers, though the ventral roots also contain some afferent fibers [70]. The dorsal and ventral roots come together in the neural foramen to form the spinal nerve as they progress distally to the periphery (Fig. 19.1); but, unlike peripheral nerves the nerve roots are not enclosed by a thick, protective epineurial sheath. The cell bodies of peripheral nerves are located in the dorsal root ganglion (DRG), which is particularly sensitive to mechanical loading, and even slight compression of normal DRGs can produce sustained neuronal activity and pain [71–73]. Direct impingement on the nerve roots and/or DRG can occur by distortion of the normal size and shape of the neural foraminae, which can occur during extreme movements of the cervical spine, stenosis, or slow osteophytic growth, or even by a herniated disc [74, 75]. The mechanics of compression- and tension-based nerve root injuries, i.e. maximum load, rate of loading, and duration of loading, contribute to the extent of pain produced after injury [76, 77]. These and other mechanical parameters that control and modulate nerve root-induced pain and pathology are further discussed in Sect. 19.4.
19.2.3 Paraspinal Musculature
Skeletal muscle constitutes much of the overall volume of the neck (Fig. 19.1), including the superficial and deep layers of muscle with attachments on the skull, shoulders, vertebrae, and spinal ligaments. Neck musculature has a dual role in neck pain – (1) by the potential for its own injury and (2) by altering the kinematics and loading of other structures in the neck that may exacerbate existing cervical spine loading and neck pain. Loading of neck musculature can lead to pain since as many as one-half of the neural units in skeletal muscle have some nociceptive function, likely through Aδ- and C-type fibers with their numerous un-encapsulated free nerve endings [78, 79].
In many neck loading scenarios, the muscles themselves can undergo concomitant contraction and elongation as a result of head or torso movement [25, 80]. For example, during whiplash, loading of the cervical musculature induces strains that exceed injury thresholds, namely in the posterior cervical muscles (splenius capitis, semispinalis capitis, trapezius); these suprathreshold strains have been reported to correlate with reports of pain in the posterior cervical region following whiplash [25, 80]. Further, after such trauma, skeletal muscles also undergo substantial structural and functional changes, including fatty infiltration, tissue degeneration, and altered activation thresholds and contraction timing, all of which can contribute to chronic pain and motor impairment [81–83]. These types of injuries can also lead to the development and maintenance of local inflammatory responses in the injured tissue that are a common peripheral mechanism for muscle tenderness and even further sensitization of nociceptors [84–86].
Though neck muscles are at risk for their own injury, they also pose a threat of injury to the other spinal tissues with which they have insertions or make direct contact. For example, semispinalis and multifidus muscle fibers insert directly on the facet capsule in the cervical spine and present a direct pathway for added loading to the capsule during muscle activation and facet joint extension [87]. Furthermore, neck muscle activation affects head and neck kinematics throughout the cervical spine, thereby also potentially altering the loading to other cervical spine tissues. Reflexive activation of neck musculature during whiplash causes significant variations in muscle extension, head displacement, and head acceleration [88]. Contraction of muscles that have their line of action along the long-axis of the spine increases axial loading in the spine and facet joints and contributes to intervertebral disc compression [25]. These altered loading states could modify the overall spine mechanics, for example potentially increasing tensile loading of spinal ligaments or disc shear, and provoking or exacerbating injury in those tissues.
19.2.4 Vasculature
The vascular components of the spine are not independently viable sources of pain, but their injury in certain loading scenarios can aggravate mechanically-induced injuries to other cervical spine tissues. The vertebral arteries supply blood to the head, brain, and neck tissues as they travel bilaterally along each cervical vertebra [89, 90] (Fig. 19.1). During certain neck loading scenarios outside physiological conditions, the vertebral arteries can become elongated, stretched, or pinched, which can potentially tear the artery [91]. For example, a vehicle occupant with a head-turned posture who undergoes a rear-end collision can undergo neck motions which cause elongation of the vertebral artery that significantly exceeds physiological tolerances [25, 92]. In particular, the vertebral artery is especially vulnerable to mechanical loading and is susceptible to pinching or tearing in the atlanto-axial region of the upper cervical spine where it passes over bony landmarks [90, 93]. Vertebral artery damage can compromise the blood supply to the brain and can also lead to secondary nociceptive pain arising from increased fluid pressure around the spinal cord [25, 94, 95]. These injuries can produce symptoms ranging from headache and neck pain to vertigo and paresthesias [94]. Given the potential mechanisms and outcomes of vertebral artery disruption, this tissue may contribute to the development of pain after injurious mechanical loading of the cervical spine. However, because of its relatively limited involvement compared to other more vulnerable and better-studied tissues, we do not focus on it in this chapter.
19.3 Pain Mechanisms
Mechanical stimulation, such as tissue loading, activates the mechanosensitive receptors that innervate the tissue to transduce the stimulus to an electrophysiological event in primary afferent fibers (Fig. 19.2). For stimuli that are above the threshold for pain sensation, the primary sensory afferents transmit nociceptive signals to the spinal cord. Normally, these nociceptive signals provide feedback to protect the organism from tissue damage or the potential for damage, so they are termed ‘physiologic’ [96, 97]. Primary sensory afferents in the PNS and higher order pathways in the spinal cord and brain convey, as well as amplify and suppress, painful sensory information [96, 98]. Potentiation of nociceptive processing leads to a state of pain hypersensitivity, or ‘sensitization’, which can become chronic or pathologic and no longer serves the physiologic purpose of pain [99, 100]. Sensitization can occur peripherally by potentiation of primary afferents, in the dorsal horn of the spinal cord, and/or supraspinally in the brain. Here, we briefly highlight the peripheral and central mechanisms of sensitization to provide a basic discussion of the pathways by which injury can lead to acute and persistent pain states. These mechanisms are reviewed in greater detail elsewhere in this book, as well as in the literature [101, 102].
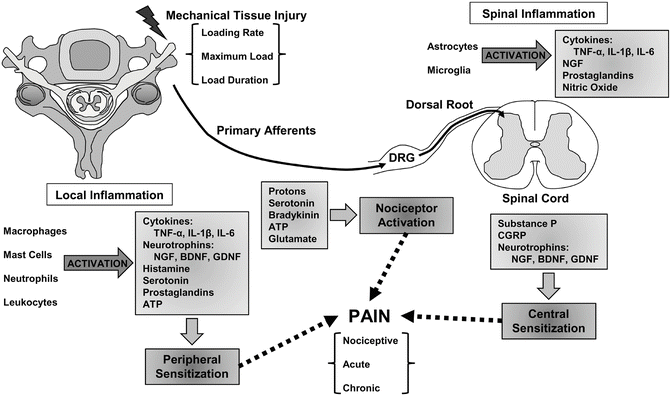
Fig. 19.2
Tissue injury induces a host of cascades in the peripheral tissues where injury occurs that also activates nociceptive afferents and can lead to central sensitization in the spinal cord and lead to pain. Nociceptor activation leads to neurotransmitter and neurotrophin synthesis in the DRG, and their transport to the spinal cord initiates spinal inflammatory cascades. Activated spinal glial cells release inflammatory mediators that further exacerbate neuronal excitability in the dorsal horn of the spinal cord. Specific characteristics of the mechanical inputs, such as loading rate, magnitude and duration, all have been shown to modulate aspects, as well as the collection, of this mechanism
Nociceptive pain is established in response to stimulus or stimuli (mechanical, chemical, thermal) that exceed the activation threshold for a given population of nociceptive fibers. Those fibers include small-diameter unmyelinated C-fibers or medium-diameter thinly-myelinated Aδ-fibers that transmit sensory signals from the periphery to the dorsal horn of the spinal cord through cell bodies in the DRG (Fig. 19.2) [78, 101, 103]. C-fibers are further classified as peptidergic or non-peptidergic, based on their characteristic expression of substance P and CGRP in the case of peptidergic fibers, and the purinergic ATP receptor, P2X3, or binding of IB4 in non-peptidergic fibers [104, 105]. Nociceptors also express receptors for a variety of neurotrophins, which are secreted factors important for neuronal growth and survival. Nerve growth factor (NGF) and glial-derived neurotrophic factor (GDNF) are neurotrophins, each of which may play an important role in pain modulation due to the presence of their receptors on peptidergic and non-peptidergic nociceptors, respectively [106].
Nociceptive pain continues only in the presence of sustained noxious stimuli [103], though pain can outlast the stimuli through peripheral sensitization, which is a potentiation of primary sensory fibers leading to lowered firing thresholds and increased firing rates once activated. Peripheral sensitization most commonly leads to decreased mechanical and thermal thresholds in the immediate area of injury [103, 105, 107]. This peripheral sensitization results from the production and peripheral release of neurotransmitters and other inflammatory factors from neurons and infiltrating non-neuronal cells in response to noxious stimuli or tissue damage [105, 108]. Nociceptors are rapidly sensitized directly via peripherally expressed receptors for mediators such as bradykinin, amines, prostanoids, NGF, and protons [108]. After injury, immune cells, including neutrophils, macrophages, and lymphocytes, initiate a cytokine cascade that may indirectly and/or directly sensitize nociceptors, further maintaining hypersensitivity [108, 109]. Peripheral sensitization includes altered nociceptive responses at the site of injury, in part as an adaptive response to promote tissue repair and healing; but, those changes can also contribute to the development of pathological pain, along with sensitization of spinal and supraspinal systems [100]. This collection of systems throughout the PNS and CNS plays a significant role in the transition from physiologic pain to the development and maintenance of persistent pain [103, 107, 110].
Persistent pain, or chronic pain, may long outlast any initiating injury event or tissue damage, and is due in large part to spinal sensitization (Fig. 19.2). Spinal sensitization is characterized by increased spontaneous activity, reduced thresholds for activation by both non-noxious and noxious stimuli, heightened responses to noxious stimuli, and sensitivity at secondary sites with no precipitating tissue damage (Fig. 19.3) [107]. Since many hypotheses have been proposed to explain the exact mechanism by which the spinal cord becomes sensitized or hyperexcitable, we only briefly highlight these theories in the context of injury; more extensive discussions are numerous in the literature [102, 105, 107, 108]. A nociceptive signal is transmitted to the spinal cord through the synaptic release of glutamate and other neurotransmitters (Fig. 19.2). Sustained nociceptive input induces rapid post-translational modifications to post-synaptic receptors that lead to increased excitability, and slower translational and transcriptional changes that promote excitability [105, 107]. Strengthening of spinal and supraspinal (i.e. brain) nociceptive signaling by changes in the ionotropic and metabotropic neurotransmitter receptors is accompanied by structural plasticity (e.g. neurite outgrowth, excitatory synaptogenesis) that may incorporate low-threshold Aβ-fibers into nociceptive pathways, causing normally non-noxious stimuli to activate the same nociceptive pathways as supra-threshold stimuli [111–114]. Ultimately, all of these responses contribute to central sensitization, though they are by no means the only mechanisms by which plasticity in the CNS modifies nociceptive pathways.
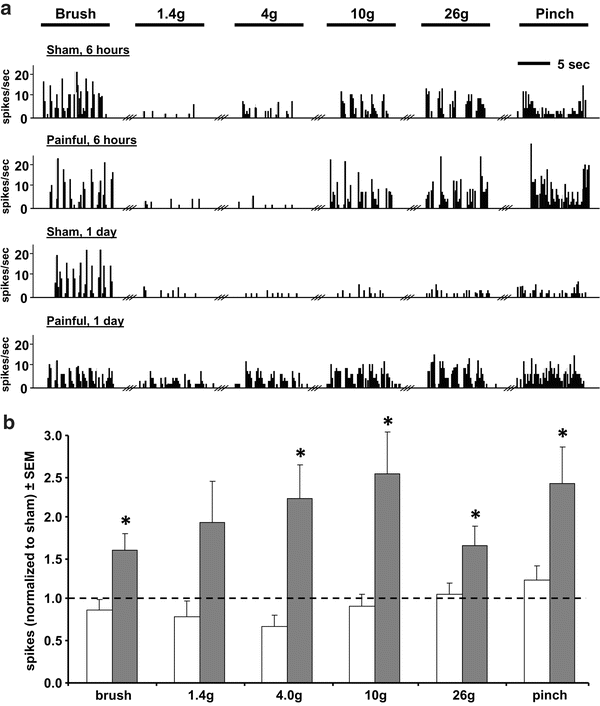
Fig. 19.3
Electrophysiological measurements of dorsal horn neuronal excitability after facet capsule injury. The evoked firing response of neurons in the C6/C7 spinal dorsal horn recorded after a painful C6/C7 facet capsule injury show increased firing over sham surgery by day 1, in response to a train of non-noxious and noxious mechanical stimuli applied to the forepaw, including light brushing, non-noxious and noxious von Frey (1.4 g, 4 g, 10 g, 26 g) filaments, and a noxious 60 g pinch. (a) Representative firing rates for neurons at 6 h or 1 day after an injury and sham procedure. (b) Spike totals for each stimulus normalized to sham illustrate the effect of injury on neuronal excitability. Evoked firing at 6 h (white bar) after a facet capsule injury was not different from sham, while firing was elevated across all stimuli by 1 day (gray bar) after sham [122]
Trophic, neuroimmune, and neuroinflammatory modulators and integrated cascades also play a role in central sensitization, just as they do in peripheral sensitization. NGF, GDNF, and a third neurotrophin, brain-derived neurotrophic factor (BDNF), each have spinal neuromodulatory roles once transported to, or released from, spinal terminals of nociceptors [115–118]. Peripheral neuroimmune and neuroinflammatory factors also activate glial cells in the CNS, including microglia and astrocytes [54]. Both types of glial cells respond to injury by changing their morphology, proliferating, and releasing several pro-inflammatory cytokines (e.g. IL-1β, TNF-α, IL-6) and mediators that further sensitize both the glial and pre- and post-synaptic spinal neuronal responses [109, 117, 119].
The pain cascades described above can be activated by mechanical loading to individual tissues or combinations of structures in the cervical spine (Fig. 19.2). For example, the primary afferent fibers that innervate the facet capsular ligament are particularly sensitive to mechanical forces generated in the capsule by distraction of the facet joint as a whole [120, 121]. Activation of those mechanosensitive afferents during injury may be sufficient to cause central sensitization by inducing excitatory changes in the neurons of the dorsal horn (Fig. 19.3) [122, 123]. Mechanical loading of the facet joint also modulates spinal glial activation and both peripheral and spinal expression of the neuropeptide, substance P, both of which are involved in the generation and maintenance of pain [124, 125]. Likewise, local injury biomechanics can affect pain by directly loading nervous tissues, like compression of the nerve root, which is known to affect neuronal structure and physiology, and induce microglial and Schwann cell proliferation and astrocytic activation at the site of injury and in the spinal cord [126–128]. The pain states experienced after mechanical injuries to both the musculoskeletal and neural tissues of the cervical spine have the potential to develop both peripherally- and centrally-mediated components and include neuropathic, inflammatory, and neuroimmune characteristics (Fig. 19.2). The relationship between injury biomechanics and pain mechanisms will be elucidated in the next section for two common cervical spine injuries – facet joint distraction and nerve root compression.
19.4 Biomechanics and Pain in Cervical Spine Injuries
As highlighted in Sect. 19.2, there are several anatomic features in the cervical spine which have been recognized as having the highest likelihood of generating pain from neck injury. Among the most vulnerable and most well-studied are the facet capsular ligament and nerve root. Moreover, both such tissues have been well characterized in their response to mechanical loading and provide interesting contrast given one is ligamentous and the other is neural tissue. With growing research efforts focused on both of these tissues, as well as their differences in mechanical responses, cellular composition and potential relevance to different types of neck injuries, we focus on each of them separately in this section. We provide clinical context for each injury modality, local and central responses to loading, and specifically discuss how the different mechanical parameters related to tissue loading/injury modulate aspects of the complicated neuroimmune cascades leading to pain (Fig. 19.2).
19.4.1 Biomechanics and Painful Facet Joint Injury
As described in Sect. 19.2 above, the cervical facet joint and its capsular ligament are among the cervical spinal tissues with great potential to generate pain when injured, particularly during whiplash [36–38]. Using anesthetic nerve blocks or provocative testing clinically, the cervical facet joint is identified as a source of pain in as many as 62 % of neck pain cases [129–131]. Facet joint injury has been hypothesized to result from two potential injury mechanisms in the joint that can lead to pain. The first produces excessive compression of the joint and impingement of the articular surfaces [132]. The articulating surfaces of the facet joints glide across one another during normal flexion, extension, or torsion of the spine [133]. Abnormal extension of the motion segment can force the bony facet of the superior vertebra to compress against the superior facet of the inferior vertebra, pinching the articular cartilage and synovial folds [132, 134, 135]. Extreme articular cartilage compression can lead to degeneration and osteoarthritis in the joint; furthermore, the synovial folds also contain nociceptive fibers and so can be a source of pain during loading [6, 136]. The second mechanism of facet joint injury includes excessive loading of the facet capsular ligament beyond its mechanical tolerance. The facet joint undergoes shear, compression, and tension during different phases of whiplash, each of which can produce tensile strain in the facet capsule [137–141]. Facet capsule strains that exceed the physiologic limit have been shown to activate mechanoreceptors and nociceptive fibers in the joint in a caprine model [120, 121].
The kinematics of the head and cervical spine during whiplash has been well defined by numerous human-volunteer and post-mortem human subject studies simulating rear-impact collisions [135, 137, 139, 142, 143]. Based on volunteer studies using high-speed X-ray imaging during low-speed rear impacts, the spine forms an ‘S’ curvature in the first 100–120 ms after impact, with the lower cervical spine undergoing extension and the upper cervical spine flexing [134, 136, 144]. The lower cervical spinal levels undergo vertebral retraction and extension, imposing tensile and shear loading to the facet joints and capsular ligaments. In particular, many studies have reported tension across the facet capsule during whiplash that induces strain levels greater than those experienced during normal physiologic motions [135, 145, 146]. In a biomechanical study using cervical spines from post-mortem human subjects, capsular ligament strains were the greatest at C6/C7, peaking at 29.5 ± 25.7 % and 39.9 ± 26.3 % strain for 6.5 g and 8 g accelerations, respectively [135]. Peak capsule strains during whiplash can be as high as five times the peak strains for the same cervical levels during normal neck bending, which have been reported to peak at 6.2 ± 5.6 % [135, 146]. Capsular strains can be further increased by more complicated out-of-plane head postures, such as a turned head [143].
Despite evidence of excessive facet capsule strains being established during whiplash [135, 143, 146], facet capsule ruptures have been reported not to occur during simulated exposures in cervical spine studies [135, 146–150]. In fact, the strains at facet capsule rupture exceed the strains experienced both during whiplash and those required for partial or minor ligament rupture [143, 151]. Furthermore, removing the contribution of the facet capsule precludes the development of behavioral hypersensitivity in animal models [125, 152]. Together, these biomechanical findings suggest that pain from facet joint injury results from sub-failure kinematics and kinetics established in the facet capsule, and that input from the afferents in the capsule is required for the establishment of pain after joint loading. This recent hypothesis is based on biomechanical, physiological and behavioral data and is contrary to prior assertions based only on biomechanical data that tissue failure is sufficient to induce such pathology.
Recently, in vivo models of facet injury have been developed that provide useful research platforms to define relationships between facet joint biomechanics, capsular loading, activation of the afferents that innervate the facet capsular ligament, CNS modifications and pain behaviors [122, 123, 153–158]. In a caprine model, distraction of the cervical facet joint produces electrophysiological activity in the afferents that innervate the joint. Specifically, mechanoreceptive fibers innervating the facet capsular ligament are activated during and for a short period (i.e. several minutes) after tensile loading of the capsule [120, 121]. Low-threshold mechanoreceptors are activated at physiologic strain levels (10–15 %) and are hypothesized to be proprioceptive [120]. A separate population of high-threshold mechanoreceptors is activated at facet capsule strains of 25–47 % [120, 121, 157], significantly higher than the strains to activate low-threshold afferents but not different from those during whiplash [135, 146, 159]. Those same magnitudes of capsular strain that activate capsule-innervating fibers are also associated with degenerative changes in the nerve fibers themselves, including localized swelling and disruption of axons, which can lead to spontaneous and neuropathic pain [160, 161]. The demonstration of activation of mechanosensitive afferents in the facet capsule during physiologic and, most notably injurious strain of the capsule, substantiates the hypothesized, but only recently proven, assertion that certain loading scenarios of the facet joint activate peripheral and central pain mechanisms that underlie clinical reports of facet-mediated pain following whiplash.
Facet capsule strains that activate afferent firing in the goat also produce persistent behavioral sensitivity in rats that mimics the primary and secondary hyperalgesia reported by whiplash and neck pain patients [135, 143, 149, 162, 163]. In a rodent model, behavioral hypersensitivity to mechanical stimulation develops in the shoulder, neck and forepaw following tensile loading of the joint that produces strains of up to 42 % in the facet capsule [155, 156, 164]. Behavioral sensitivity begins by 1 day and is sustained for up to 6 weeks after joint loading [165]. The onset of mechanical sensitivity after injury is indicative of both hyperalgesia, or pain hypersensitivity, and allodynia, which is an increased pain response to non-noxious stimuli [3]. Behavioral sensitivity at sites remote from the injury, like the forepaw, is termed ‘secondary hyperalgesia’ and is a key clinical indication that the pain may be driven at least in part by central sensitization, or the potentiation of nociceptive elements located in the dorsal horn of the spinal cord [107]. Behavioral sensitivity is caused by the same magnitude of facet capsule strains that activate afferent fibers in the capsule; this suggests that the initial biomechanically-induced electrophysiological activity in the facet capsule may activate both peripheral and central pain mechanisms that result in primary and secondary hyperalgesia.
Facet capsule injury induces electrophysiological changes in the dorsal horn of the spinal cord that are consistent with those of central sensitization, including increased spontaneous activity, reduced thresholds for activation by both non-noxious and noxious stimuli, and heightened responses to noxious stimuli. As early as 1 day after injury, neurons in the dorsal horn at the same spinal level as the injury develop hyperexcitability in response to both innocuous and noxious mechanical stimuli at the forepaw (Fig. 19.3) [122]. That hyperexcitability is sustained for at least 7 days after the initial capsule injury [123]. Neurons also exhibit increased spontaneous activity and afterdischarge, as well as a functional shift of secondary neurons in the dorsal horn from low-threshold mechanoreceptors (LTMs) or nociceptive-specific (NS) neurons to wide dynamic range (WDR) neurons [123]. Although LTMs are normally activated only by light mechanical stimulation and NS neurons only by nociceptive stimuli, WDR neurons are responsive to a large range of stimuli, including both innocuous and noxious stimuli [96]. A gain-of-function shift of LTMs to WDR neurons can produce increased amplification of noxious stimuli in the CNS. Likewise, a shift of NS neurons to WDR neurons can activate nociceptive pathways by normally non-noxious stimuli, leading to decreased pain thresholds and behavioral hypersensitivity [107]. Electrophysiological changes such as those that occur after facet joint injury are likely driven by both peripheral (facet capsule, afferent fiber, DRG) and spinal cellular cascades that ultimately increase neuronal excitability.
Facet joint injury induces a host of cellular-level changes in the peripheral and central nervous systems that contribute to increased neuronal activity and ultimately pain. Many of these changes directly, and indirectly, modulate elements of the glutamatergic system, which is the primary excitatory neurotransmitter system in the spinal cord [154, 158, 166]. For example, facet joint injury upregulates protein kinase C epsilon (PKCε), a regulator of glutamate release and potentiation of ionotropic glutamate receptor NMDA [166–168]. The metabotropic glutamate receptor, mGluR5, which is important for enhanced nociceptive signaling in dorsal horn neurons [169–171], is also upregulated in the DRG and spinal cord as early as 1 day after facet capsule injury [158]. Further, upregulated mGluR5 can contribute to astrocytic activation and enhanced PKCε activity, suggesting another potential mechanism by which spinal neuronal excitability may become increased after painful facet capsule loading [158, 172, 173]. Facet capsule strains induced by whiplash-like joint loading can also disrupt the ability of capsule-innervating afferent fibers to produce substance P mRNA for at least 1 week after injury [124]. At the same time, substance P protein levels are increased in the DRG and decreased in the spinal cord over the same time frame [124], suggesting that joint injury may induce axonal dysfunction that affects protein utilization and retrograde transport along afferent fibers. The effects on cellular function that are induced by painful joint loading extend also to spinal glial cells, including increased spinal astrocytic activation and reactive synthesis and release of pro-inflammatory cytokines in both the DRG and spinal cord [125, 164, 174–176]. The integrated neuronal and astroglial cascades that have been shown to be activated after painful facet capsule injury collectively contribute to a general increase in excitatory signaling in the dorsal horn that can manifest as peripheral and central sensitization observed after mechanical facet joint injury.
Biomechanical evidence supports that a range of capsular ligament sub-failure strains can generate pain and contribute to sustained sensitivity in association with a host of cellular nociceptive responses. For this particular tissue injury, animal models have provided valuable platforms to study the effects of biomechanical loading on the initiation of pain-related electrophysiologic, immunologic, inflammatory, and neurophysiologic changes in both peripheral and central nervous system structures. Further, such systems have allowed the temporal and spatial definition of an integrated schema from the initial injury spanning the development of persistent pain (Fig. 19.2). Continued integrated biomechanical and physiologic research is needed to relate the findings in animal models to injury scenarios in humans, which are currently informed by simulation studies and clinical reports, but which lack the ability to incorporate cellular and molecular level assays. Although injury to the cervical facet joint is a major contributor to whiplash-associated pain, other anatomical structures in the cervical spine are vulnerable to mechanical insult during different loading scenarios. Of note, the cervical nerve roots, which will be addressed in the next section, are highly susceptible to compression- and tension-based loading and contribute to pain based on the magnitude, duration and rate at which they are deformed.
19.4.2 Biomechanics and Painful Nerve Root Injury
As described above, nerve root compression is a common injury in the cervical spine and leads to a host of pain states, which depend on the mechanical profiles of the injury (Figs. 19.1 and 19.2). The clinical syndrome of radiculopathy originates after nerve root injury that can be induced by transient or sustained compression or tension, chemical stimuli, or a combination of both [177–179]. Cervical radiculopathy is characterized clinically as pain, paresthesia or hypersensitivity to mechanical and temperature stimuli radiating down the arm [180–183]. Although both mechanically- and chemically-induced injury to the cervical nerve roots have been confirmed to contribute to the development of chronic radicular pain [184–186], we focus on mechanically-induced neural trauma and its contribution to nerve root-mediated pain in the cervical spine. Mechanical trauma to the nerve roots can occur due to a number of diseases, disorders, and/or spinal loading scenarios. Direct compression, or associated tethering, of the roots can be induced slowly over an extended period of time which is often associated with foraminal narrowing that can occur with the natural progression of spinal aging or degeneration. In contrast, traumatic injury to the nerve root tissue can result from rapid increases in high-magnitude loads or deformations that can be produced by injuries, such as a herniated disc protruding into the intermedulary cavity or spinal trauma causing the intervertebral foramen to become obstructed [180, 187].
Pain induced by nerve root loading can be sustained long after the removal of the tissue insult. The specific mechanics of injury, including the type of load (i.e. compressive or tensile), the loading rate, the maximum load, and the duration of loading all contribute to the resulting behavioral sensitivity (Fig. 19.2) [53]. For example, one such modulator is the duration of root compression, which can be only transient in some injury conditions, including sports or automotive related traumas, or sustained due to disc herniation and spondylosis [180, 188–191]. This discrepancy has also complicated the clinical understanding of such injuries and their associated pathologies because a transient compression may not be visible by conventional imaging. Consequently, the resulting neck pain would not be attributed to a mechanical injury to the nerve root. Animal models of mechanical root injury have been instrumental in enabling the development of a better understanding of the cellular and molecular mechanisms that are initiated after nerve root trauma and how they contribute to the development of acute and/or chronic pain. Although the effects of nerve root compression profiles have been the focus of more investigations than those for tension, both injury modalities have been shown to play a role in pain.
Using animal models, the rate at which tension is applied to the nerve root and the maximum tensile strain have been shown to alter both the mechanical response of the neural tissue and the physiologic function of the neurons that are undergoing the tensile loading [192, 193]. Nerve roots tested in tension to failure exhibit a rate dependence to their mechanical strength at failure; a dynamic loading rate (15 mm/s) results in greater mechanical parameters than those for quasistatic loading (0.01 mm/s): maximum load (13.9 ± 7.5 versus 5.7 ± 2.7 g), maximum stress (624.9 ± 306.8 versus 257.9 ± 111.3 kPa), and elastic modulus (2.9 ± 1.5 versus 1.8 ± 0.8 MPa) [192]. This rate-dependent response is not surprising as it has been commonly demonstrated for other soft tissues [194, 195]. Of interest though, that study also reported that the magnitude of tensile strain applied to the root also affects measures of the tissue’s physiologic function, as assessed by electrically-evoked compound action potentials [193]. Tensile strains between 10 % and 20 % were sufficient to decrease the conduction velocity and action potential amplitudes compared to strains less than 10 % [192]. Further, strains greater than 20 % completely blocked action potential conduction irrespective of the strain rate [193]. Although these studies measured structural and functional outcomes in the lumbar nerve roots of the rat during tensile injury, similar tension-based painful injuries occur in the cervical nerve roots and the pain outcomes are hypothesized to have a similar dependence on the mechanics of injury.
As with modeling the facet joint injury, animal models of painful nerve root compression have defined the pathophysiological consequences of root compression and in some cases can relate the tissue injury to those mechanisms in the context of pain. There is extensive literature on the role of nerve root (and cauda equina) compression in mediating low back pain that has formed the basis of most of the work in the cervical spine [4]. More severe nerve root compressions (i.e. higher loads, greater deformation, longer compression times) produce a more-robust behavioral sensitivity response [76, 196–200]. In fact, a transient cervical nerve root compression (applied for 15 min) in the rat using a compressive load of 26 mN elicits mechanical allodynia for only 24 h whereas compressive loads at or above the higher (38 mN) load applied to the nerve root for the same duration produces mechanical allodynia that is sustained for at least 7 days after injury [201]. This suggests a load threshold exists for root compression to induce pain that is sustained. In the same rat injury model, a load of 98.1 mN, which is even further above that load threshold when applied for 15 min, does not initiate mechanical or thermal sensitivity when it is applied for a duration of only 3 min [76, 202, 203]. The degree of tissue impingement or disruption produced by the compression is yet another mechanical parameter that modulates nerve root-induced behavioral sensitivity. For example, greater lumbar nerve root compressions (with an average strain of 39 ± 7 %) also produce more robust mechanical allodynia responses over a 14 day postoperative period compared to those compressions inducing lower strains of 21 ± 5 % [198]. Although that study relates the initial nerve root strain to the pain outcomes, the degree of nerve root deformation that is maintained after the removal of a transient compression has also been related to the resulting behavioral hypersensitivity [76]. Cervical roots compressed for 30 s or 3 min recover to over 88 ± 5 % of their original width and do not produce mechanical allodynia. In contrast, nerve roots compressed for 15 min recover to only 72 ± 13 % and do induce mechanical allodynia [76]. Together, these studies suggest disruption of the overall structural integrity of the nerve root during and after its compression may directly contribute to the onset and maintenance of nerve root-mediated pain.
Compression of the nerve root applies direct compression to the afferent fibers within the root and induces a wide array of phenotypic and electrophysiological changes at the location of compression (Fig. 19.2). Biochemical and cellular changes are also initiated at locations remote from the injury site, including in the cell bodies of the compressed afferents (located within the DRG) as well as in neurons and glia within the spinal cord with the release of cytotoxic amounts of neurotransmitters and cytokines at the synapses of the injured afferents [107, 204]. As with nerve root-mediated behavioral sensitivity, cellular changes induced by painful root compression also depend on the mechanics of the primary initial tissue compression [76, 77, 196, 197, 200, 202, 203]. The biomechanical changes that occur within cells at the primary site of injury and at remote locations provide the link between the specific injury mechanics (i.e. maximum load, rate of loading, and load hold time) and nerve root-induced behavioral sensitivity.
The magnitude of root compression contributes to changes in that tissue’s structure through the degeneration of its constituent neurons and the associated infiltration of immune cells, and also modulates levels of neuropeptides that are expressed by the compressed and/or injured neurons in the root. For a painful cervical root compression in the rat that lasts 15 min a load of 32 ± 9 mN is sufficient to reduce expression of neurofilaments in the neurons in the injured nerve root by 1 week after injury, indicating their degeneration at this time point [201]. This load threshold is similar to the load threshold that also induces persistent behavioral sensitivity (38 mN) [205], suggesting that disruption of neuronal structure in the nerve root after its compression is associated with the maintenance of pain. In addition the magnitude of compression load also modulates the expression of neuropeptides in the DRG; the number of small diameter nociceptive afferents in the DRG that express substance P decreases by day 7 after a 15 min compression proportionate to the peak compressive load applied to the root [196, 201, 205]. This decrease in substance P in the DRG may be attributed to an increase in the synaptic release and utilization following painful root compression and may also contribute to the subsequent behavioral sensitivity [205, 206]. Yet, a compressive load of 20 ± 10 mN, which is below the load thresholds for producing sustained mechanical allodynia and neuronal degeneration, is sufficient to induce macrophage infiltration in the nerve root at this time, which indicates that macrophage infiltration at the root may not directly depend on the load magnitude [201]. Although macrophage infiltration at the root 7 days after its compression does not appear to be directly related to the presence of behavioral sensitivity, by day 14, when pain is still present, infiltrated macrophages exhibit a phagocytotic morphology, aiding in the removal of myelin debris from degenerated neurons [126]. This response suggests that non-quiescent macrophages at the nerve root may be involved in prolonging pain after nerve root compression. Taken together, this collection of studies suggests that afferent responses, including their degeneration and altered transport of substance P, is controlled at least partially by the magnitude of compression and dictates nerve root-mediated pain at this time point.
In addition to the magnitude of the applied load, the duration of compression mediates the disruption of neuronal structure and function. A nerve root compressed for a short period (30 s or 3 min) at 98.1 mN, which is above the load threshold for producing sustained behavioral sensitivity when applied for 15 min [196], exhibits almost full recovery of its original shape (over 88 ± 5 % of the original width of the root) [76]. This suggests that despite the magnitude of load being painful if applied for a certain time period, if the same load is applied for a short duration there is insufficient time for the cytoskeletal structures to become sufficiently deformed and/or undergo injury at the axonal level. That study suggests that a critical duration of compression exists between 3 and 15 min in which the disruption in root structure remains after the compression is removed. That same duration is also necessary to induce immediate changes in electrophysiological responses by the compressed neurons where they synapse in the spinal cord. During a transient 98.1 mN compression, mechanically-evoked action potentials are significantly reduced in the spinal cord of the rat at 6.6 ± 3.0 min of applied compression [207]. Since the duration of nerve root compression required to develop sustained pain for this 98.1 mN load is also between 3 and 10 min [202], the sustained disruption of axonal structure may influence the propagation of action potentials from the nerve root to the spinal cord and contribute to the initiation pain.
The rate at which nerve roots are compressed also mediates the consequences on neuronal function, and therefore, may also contribute to behavioral sensitivity just as load magnitude and duration have modulatory effects. In a porcine model of nerve root compression, Olmarker et al. [208] was among the first to demonstrate that faster rates of root compression (0.05–1 s to reach the maximum load) reduce neuronal conduction velocities in the compressed nerve root compared to slower rates (20 s to reach that same maximum load) [208]. Due to the viscoelastic nature of neural tissue like the nerve root, as the loading rate increases the peak load decreases. This has been demonstrated for rodent nerve root tissue during its compression; a 0.65 mm compression applied at a rate of 0.004 mm/s generates a peak reaction load of 102.3 ± 31.8 mN, which is significantly lower than the peak load of 177.9 ± 63.5 mN when the root is compressed the same amount but at a dynamic rate of 2 mm/s [205]. This study highlights the dependence of the mechanical parameters of nerve root compression on each other, suggesting that the peak load, rate of loading, degree of deformation and hold time must all be considered when defining the role of compression mechanics in nerve root-mediated pain.
As mentioned in Sect. 19.3 on pain mechanisms, the mechanics of nerve root compression also regulate neuronal signaling in the spinal cord where the injured afferents synapse (Fig. 19.2). The fact that evoked action potentials in the spinal cord are reduced (or abolished) at approximately 6 min of compression by a 98.1 mN load [207] indicates that a disruption of neuronal signaling occurs during the injury itself. Alterations in neuronal signaling are also present in the spinal cord at 1 week after root compression [196]. Spinal expression of the neuropeptides, substance P and CGRP, is decreased on day 7 after a painful compression of 98.1 mN for 15 min [196]. Of the two peptides, only CGRP expression depends on the magnitude of load [196], but the load threshold necessary to induce changes in that peptide was below that necessary to induce persistent behavioral sensitivity (19.5 and 38.2 mN, respectively) [196, 201]. Relatedly, the extent to which the spinal glutamatergic system is modified after nerve root compression in the rat depends on the magnitude of applied load, with increased expression of the glutamate receptor mGluR5 evident on day 7 [203]. However, spinal mGluR5 expression is elevated after a load of 558.6 mN compared to 98.1 mN, despite the absence of different behavioral sensitivity responses at the same time point [203]. These findings indicate that the modulation of biochemical factors other than CGRP and mGluR5 in the spinal cord may play a role in compressive load controlling the production and maintenance of behavioral sensitivity and that different responses may be mediated by different mechanical factors.
The magnitude of compression also controls the extent of spinal astrocyte activation, and does so in parallel with behavioral responses [76, 197, 200]. In contrast to biomarkers such as mGluR5 which exhibit load dependence even when behavioral outcomes do not [203], compressive loads of 98.1 and 588.6 mN, which result in similar levels of persistent mechanical allodynia also induce similar degrees of spinal astrocyte activation [197, 203]. This implicates activated spinal astrocytes in partially contributing to the load dependence seen with persistent behavioral sensitivity after root compression. Spinal astrocyte activation is not observed on day 7 after shorter compressions (30 s, 3 min) that also do not produce mechanical allodynia, but is evident after longer compressions (10 or 15 min) [76, 202]. Interestingly, the longer duration compressions are above that required to reduce neuronal firing in the spinal cord (6.6 ± 3.0 min) [207] and correlate to durations that also do not allow for sufficient structural recovery in the nerve root after the compression is removed [76]. Taken together, this collective group of studies implies that mechanical parameters of compression may immediately modulate axonal firing in the spinal cord and structural recovery of the nerve root which may further be involved in spinal astrocyte activation contributing to the maintenance of nerve root-mediated pain (Fig. 19.2).
19.4.3 Other Anatomic Sources of Mechanically-Induced Neck Pain
This chapter has primarily focused on pain originating from mechanical injury to the cervical facet joint and nerve root because of the strong history of engineering research related to these tissues, as well as the detailed understanding of pain physiology. Nonetheless, mechanical loading of other anatomic sources, such as the spinal cord, intervertebral disc and paraspinal musculature, of the cervical spine does occur and also contributes to neck pain (Fig. 19.1).
Traumatic spinal cord injuries (SCIs) can initiate persistent pain along with the other host of pathologies that are far more debilitating; a review of SCIs is beyond the scope of this chapter and can be found with extensive discussion elsewhere in the literature [209–213]. The mechanical trauma experienced by the spinal cord during incomplete spinal cord injury induces neuronal death and degeneration and glial scarring within the spinal cord, which contribute to persistent neuronal dysfunction and pain [214]. These cellular and molecular changes occur at the same spinal level as the injury, also known as primary or ‘at-level’ responses, and also at spinal levels above and below the injury, which are known as ‘secondary’ injury responses [211, 213, 215]. Pain from SCI has an immediate onset and is believed to be caused by the hyperexcitability of injured afferents themselves [211]. Secondary pain develops gradually and may be due to the widespread activation of glial cells that is induced by a host of pathological processes including microvascular hemorrhage, ischemia, edema, free radical-induced oxidative stress and excitotoxicity [211, 214, 216]. The extent of spreading of these secondary responses after SCI depends directly on the energy delivered to the neural tissue during injurious impact and increases the extent of pain [214, 216]. Similar to the pain induced by mechanical injury to the facet capsule and/or nerve root, the cascade of CNS responses that is initiated following spinal cord injury includes neuronal (chemical and anatomical) and inflammatory components that together contribute to pain (Fig. 19.2) [213].
Neck pain can also originate from injury to the intervertebral disc or nearby musculature since these structures are also innervated by nociceptive neurons (Fig. 19.1) [11, 14, 78, 79, 217]. Similar to the vulnerability of the facet joints to injury during certain neck movements, the cervical discs are also at risk for excessive compression, tension and shear during neck motions occurring during traumatic events which only increases as the disc degenerates with age [218, 219]. However, cervical disc injuries can be caused by more severe loading scenarios that involve multiple tissues [220] and are frequently accompanied by concomitant injuries, such as intervertebral canal narrowing and gross anatomical disruption and instability of the cervical spine, which themselves can also contribute to pain as discussed above [221]. Although sub-failure muscle injury (e.g. sprains) in the cervical spine can induce pain [80, 222, 223], it is often short-lived and does not become chronic [25, 84]. Yet, as with disc injuries, injuries to the cervical musculature can result in spinal instability which can affect joint loading and amplify pre-existing pain states [83, 86, 88].
19.5 Experimental Techniques to Study Pain Biomechanics
Although most areas of contemporary injury biomechanics require the integration of engineering approaches to define the mechanics with the physiological sequelae of tissue loading, pain biomechanics, in particular, requires experimental techniques which incorporate both such methodologies for a complete understanding of how tissue injury mediates the function or dysfunction of tissues, cells, and molecular cascades. With the advent of more sophisticated technologies, with improved spatial and temporal resolution, it has become possible to make mechanical measurements in different model systems – ranging from the human to the isolated cell – and to relate those mechanics to cellular function/dysfunction beyond those of mechanical function.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


