Fig. 9.1
The factors that affect to GLUT4 translocation and glucose utilization in the skeletal muscle
The working mechanism of insulin resistance is complex, probably involving numerous factors, including defective tyrosine phosphorylation of IRS-1. IRS-1 is phosphorylated on a tyrosine residue for signal transduction to the downstream pathway. More than 50 phosphorylation sites exist on serine/threonine residues and hinder the phosphorylation of tyrosine residues. C-Jun N-terminal kinase (JNK) and nuclear factor-kB (IkB) are representative serine/threonine kinases for IRS-1 [14]. At first, JNK is activated by TNFα, and then JNK phosphorylates serine 307 of IRS-1, which suppresses phosphorylation of the major tyrosine residue of IRS-1 and subsequently attenuates insulin signaling [14]. Further, when S6kinase (S6K) is activated by insulin and amino acids, phosphorylation of serine 636/639 of IRS1 is accelerated, leading to attenuation of insulin signaling [15]. Phosphorylation of IRS-1 serine/threonine residues inhibits binding to PI3-K and accelerates proteasomal degradation of IRS-1 [16]. However, recent detailed analysis of the IRS phosphorylation sites suggests that serine/threonine phosphorylation does not necessarily lead to negative regulation of insulin signaling and that, in contrast, insulin signaling may be enhanced depending on the combinations of different phosphorylation sites. Under diverse physiological/pathological conditions, IRS-1 appears to modulate insulin signaling by transmitting the message for enhancing or suppressing insulin signals [17].
The findings available so far concerning how Class IA PI3-K is involved in insulin resistance are confusing [18]. PI3-K consists of a catalytic subunit with lipid kinase activity and a regulatory subunit that binds to the phosphorylated tyrosine residue of IRS. In mammals, the regulatory subunit is encoded by three genes, namely, pik3r1, pik3r2, and p55PIK [19]. While pik3r1 encodes three splice variants (p85α, AS53, and p50α), pik3r2 and p55PIK encode p85 and p55PIK, respectively [19]. Short isoforms, AS53, p50, and p55PIK, are expressed in limited cells or tissues while p85α and p85β are ubiquitously expressed, though expression of p85α is considered significant. In the skeletal muscle of patients with type 2 diabetes, the expression of these regulatory subunits increases [20]. Surprisingly, even in p85alpha [21] and p85beta [22] knockout mice, insulin sensitivity can be strengthened. It is difficult to fully explain these results. It is possible that p85 plays an essential role in insulin signaling by guiding P13-K toward IRS, though excess p85 suppresses insulin signaling. Phosphatase and tensin homolog (PTEN) deleted on chromosome ten is a phosphatase that antagonizes P13-K by dephosphorylating PIP3. It was difficult to induce insulin resistance in the skeletal muscle-specific PTEN knockout mice with a high-fat diet [23]. Therefore, insufficient regulation of the PIP3 production by PI3-K and PTEN may cause insulin resistance.
Protein-Tyrosine phosphatase 1B (PTP1B) is a phosphatase that dephosphorylates tyrosine residues of IR and IRS. Expression of PTP1B is increased in human insulin-resistant skeletal muscle [24]. Insulin sensitivity increases in skeletal muscle-specific PTP1B knockout mice and protects the body from high-fat diet-induced obesity and diabetes [25] and from TNFα-induced insulin resistance [26]. Based on these findings, PTP1B is being considered as a therapeutic target for treating insulin resistance.
9.3 Ectopic Fat
Males and postmenopausal women do not have sufficient estrogen, which functions to facilitate the accumulation of subcutaneous fat; therefore, visceral fat is easily accumulated. When this fat reaches a certain level, the energy that can no longer be stored as visceral fat must accumulate in the skeletal muscle and the liver, which do not primarily function as sites of fat storage. This accumulated fat can be consumed as a source of energy, but when the fat remains in these tissues for an extended period, insulin resistance intensifies. Asian populations, including the Japanese, have a low capacity for accumulating subcutaneous fat and instead have a tendency to accumulate ectopic fat; therefore, they are predisposed to diabetes. Accordingly, ectopic fat, along with visceral fat, should be strictly monitored.
We do not fully understand how the lipid in the skeletal muscle induces insulin resistance. However, new findings rule out the traditional explanation, and new hypotheses have been developed (see a review by Sulman for a detailed explanation [27]). According to the traditional theory, ectopic lipids accumulate in skeletal muscle and fatty acid oxidation increases. This causes the concentration of mitochondrial acetyl-coenzyme A to increase, which is followed by the following sequence of events: supression of pyruvate dehydrogenase (PDH), inhibition of glucose oxidation and intracelluar citrate concentration, suppression of phosphofructokinase (PFK), increase in intracellular glucose-6-phosphate concentration, suppression of hexokinase (HK) activity, increase in intracellular glucose concentration, and suppression of glucose transport (the so-called Randle’s hypothesis [28]). However, according to recent reports, the concentrations of intracellular glucose-6-phosphate and glucose are in fact reduced by lipid accumulation. Based on this result, the following alternate hypothesis was proposed: the increase in intracellular diacylglycerol (DAG) concentration activates PKCθ, serine 1101 of IRS-1 is phosphorylated, and insulin signaling is suppressed.
The author’s group performed three different lipidomics analyses in rats comparing the effects of a 10-week running exercise regime and a 16-week high-fat diet to determine the difference in the profiles of lipid species in the skeletal muscle [29]. The authors found that DAG content was not affected by either exercise or by high-fat diet, while insulin sensitivity of skeletal muscle was increased by exercise and suppressed by high-fat diet. These results indicate that DAG may not be the only causal agent in insulin resistance. Further, linoleic acid-containing phosphatidylcholine, sphingomyelin, and docosahexaenoic acid-containing phosphatidylcholine were found to be chronic exercise-induced lipids. In contrast, arachidonic acid-containing phosphatidylcholines, phosphatidylethanolamines, and phosphatidylinositol were found to be high-fat diet-induced lipids. In addition, minor sphingomyelin, which has long-chain fatty acids, was identified as a high-fat diet-specific lipid. There are numerous lipid species in the skeletal muscle, and their physiological activities vary. Therefore, a detailed analysis of the behaviors of these lipid species by mass spectrometry or by other methods would further delineate the mechanisms that lead to insulin resistance.
9.4 Effect of Exercise (Muscle Contraction) on Glucose Transport in the Skeletal Muscle
The importance of exercise in the control of blood glucose has been widely recognized. Dynamic contraction of large muscle groups, even for a relatively short time, promotes glucose uptake into the skeletal muscle. When the skeletal muscle from rodent hind limb was cultured in the presence of oxygen and electrically stimulated to induce contraction, glucose uptake occurred, as in the case of insulin stimulation [30]. Glucose uptake in response to muscle contraction has the same effect as glucose uptake in response to insulin, because GLUT4 translocation eventually occurs, though the signal transduction pathway to the final step is different. Even in insulin-resistant patients with type 2 diabetes, normal glucose uptake into the skeletal muscle can often be maintained by exercise, as long as their condition is not serious [31]. This is because of the activation of different signal transduction pathways by exercise and by insulin. This difference can be demonstrated by a classical experiment in which muscle contraction-induced glucose uptake is not suppressed by the PI3-K inhibitor [32–34]. Further, in the skeletal muscle of IR knockout mice, normal muscle contraction-induced glucose uptake is maintained [35]. In general, there are some factors that can evoke or intermediate exercise- and contraction-induced intracellular signaling (Fig. 9.2). Nevertheless, the identity of the specific intracellular molecules that are involved in this process remained unknown for a long time. A major breakthrough occurred when the role of AMP-activated protein kinase (AMPK) was discovered [30].
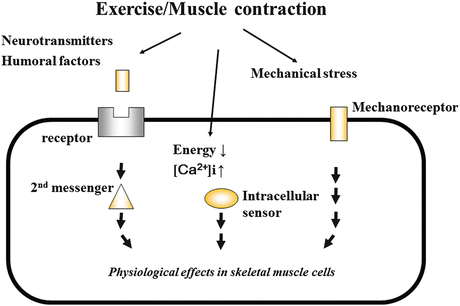
Fig. 9.2
Intracellular signaling evoked by exercise and muscle contraction
AMPK is the serine/threonine kinase that is activated when intracellular energy decreases [30]. AMPK is a heterotrimer consisting an α subunit, which contains the catalytic domain; a γ subunit, which mediates binding to ATP/ADP/AMP; and a β subunit, which links the other two subunits and contains a glycogen binding domain [36]. Kinase activity is enhanced or suppressed when AMP or ATP, respectively, binds to the γ subunit. When AMPK is activated, catabolic action is enhanced for energy production, while anabolic action for energy consumption is concurrently suppressed, so that the intracellular energy level increases. In summary, upon AMPK activation, glucose and fatty acid uptake, glycolysis, and oxidation are promoted, whereas the synthesis of lipids, glycogen, and protein is suppressed. A number of studies into the regulation of muscle contraction-associated glucose uptake were the starting point for understanding the multiple physiological activities of AMPK.
At the time of muscle contraction, the intracellular energy level goes down while the ratio of AMP to ATP increases. Subsequently, AMPK is activated, and an intracellular signaling cascade for GLUT4 translocation is initiated. AMPK phosphorylates downstream TBC1 domain family member 1 (TBC1D1) [37] and member 4 (AS160) [38] and suppresses the GTP hydrolytic enzymes of these proteins. These molecules become active when dephosphorylated. Usually activated in the state of abundant intracellular energy, these molecules suppress the downstream Ras homologous from brain (Rab) and prevent the translocation of GLUT4 to cell membrane. Rab8A, Rab13, Rab14, and other Rab proteins are known to be involved in GLUT4 regulation in the skeletal muscle [4], but additional molecules may be identified in the future. The GTP-bound and GDP-bound forms are the active and inactive forms of Rab, respectively. Therefore, when TBC1D1 and AS160 are phosphorylated and the GTPase activity is inactivated, the GTP-bound, active form of Rab increases and accelerates the translocation of GLUT4. Further, upon phosphorylation, TBC1D1 and AS160 bind to 14-3-3 and are prevented access to Rab [39].
A recent study suggests that sucrose nonfermenting AMPK-related kinase (SNARK) is an upstream factor involved in inactivation of TBC1D1 and AS160 [40]. SNARK, a member of the AMPK-related kinase family, is a serine/threonine kinase that, like AMPK, is phosphorylated by LKB-1. However, since LKB1 is constitutively activated, the status of the downstream target molecule likely determines whether the signal is transmitted. For instance, the site at which LKB-1 phosphorylates AMPK is exposed when AMPK is in the AMP-bound state (when the intracellular energy is low). Phosphorylation of threonine at this site works as a switch for the activation of AMPK. In contrast, in the case of SNARK, it is not known which signal, for example, a change in the cell environment or the presence of a second messenger, is a trigger for phosphorylation by LKB-1. In either case, AMPK and SNARK are the molecules that transmit the glucose uptake signal induced by muscle contraction. AMPK is stable as a heterotrimer of the α, β, and γ subunits. When the α-γ linking β subunit is missing, the trimer cannot be formed, leading to collapse of AMPK. In AMPK β1/β2 double knockout mice that lack the two β subunit isoforms (β1, β2), the muscle contraction-induced glucose uptake is reduced to approximately half [41]. A similar reduction is found in the LKB-1 knockout mouse [40]. Based on these findings, there may be other signaling molecules besides AMPK and SNARK that are associated with muscle contraction-induced glucose uptake.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
 Mechanism of Skeletal Muscle Contraction: Role of Mechanical Muscle Contraction in Glucose Homeostasis
Mechanism of Skeletal Muscle Contraction: Role of Mechanical Muscle Contraction in Glucose Homeostasis
 Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
 Fracture Risk in Diabetes
Fracture Risk in Diabetes
 Body Temperature Regulation During Exercise Training
Body Temperature Regulation During Exercise Training
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


