Perception of pain
Pain tolerance
Pain referral
Cutaneous and visceral pain
Sharp, pricking, burning, and/or cutting sensations
Easily or moderately tolerated
No pain referral and no secondary hyperalgesia
Musculoskeletal pain
Aching and cramping
Poorly tolerated
Often accompanied by pain referral and secondary hyperalgesia
Musculoskeletal Pain Associated with Muscle Injury
Pain arising from damaged muscle is classified as either acute or chronic. Macro- or microtrauma, along with mechanical overload of muscles during contraction or stretch phases, is the most common cause of acute musculoskeletal pain [13]. Common muscle soreness, or type I muscle strain, arising from mechanical overload or overuse experienced following exercise or work-related actions is typically perceived during or immediately following activity. The duration of this type of musculoskeletal soreness is transient, generally lasts for only hours, and is coupled with muscle stiffness and tenderness [13, 14]. Distinct from the symptoms associated with type I muscle strain, delayed onset muscle soreness (DOMS) describes musculoskeletal pain that develops up to 24 h after overactivity or muscular overload. As DOMS progresses, musculoskeletal pain, weakness, and stiffness steadily increase in intensity and peak within 2–3 days following onset. Due to underlying cellular damage and ongoing inflammatory processes (discussed later in this chapter) related to the acute muscle damage associated with DOMS, full recovery of muscle function may take weeks to occur [14].
The proximal causes underlying chronic musculoskeletal pain following overload or overuse are somewhat more difficult to identify. Often, it is the accumulation of muscle trauma or repeated acute muscle damage that leads to the onset of chronic musculoskeletal pain which is often accompanied by hyperalgesia [13]. There are several musculoskeletal and neuromuscular disorders (most outside the scope of this chapter) that include symptomatic chronic muscular pain. Two disorders with associated chronic musculoskeletal pain are described below .
Musculoskeletal Pain Associated with Myofascial Pain Syndrome
Both hyperalgesia and referred pain of muscular origin are hallmarks of a musculoskeletal pain condition known as myofascial pain syndrome (MPS) and highlight the unique nature of the myosensory pathway. MPS is caused by the appearance of myofascial trigger points (TrPs) located within muscles. Activated by chronic or acute muscular overload or overuse, TrPs are defined as “taut bands” of muscle fibers that extend the length of the entire muscle and are chronically contracted [15–18]. Interestingly, the constant contracted state of these taut bands is independent of typical presynaptic motor neuron activity and subsequent end-plate action potential generation. Instead, chronic contraction of the taut bands seems to originate via abnormal acetylcholine release and subsequent extremely high-frequency miniature end-plate potential activity [15–18]. Activated TrPs, which create multiple taut bands within a muscle or muscles, subsequently lead to the formation of areas known as zones of tenderness which are associated with regions of hypoxia. It is the creation of these zones of tenderness that gives rise to sensory musculoskeletal pain associated with MPS. The area of TrP formation becomes hypersensitive to stimulation and normally nonpainful stimuli become painful (allodynia). The immediate consequence of TrP formation in an affected muscle is increased fatigability, pain during contraction, and a restriction of range of motion . Additionally, the pain associated with TrP formation is referred to other muscle groups, distinct from the locally affected muscle [15–18]. Progressive musculoskeletal pain associated with spreading hyperalgesia is a common occurrence associated with MPS due, in part, to TrP formation in compensating muscles or abnormally contracted postural muscles [16]. Later in this chapter, we will discuss potential mechanisms which may underlie the activation of nociceptive systems which transmit both proximal and referred pain stimuli associated with MPS .
Musculoskeletal Pain Associated with Fibromyalgia
It is important to distinguish the terminologies associated with the underpinnings of musculoskeletal pain associated with fibromyalgia and contrast them with those that describe the origins of pain related to MPS. Fibromyalgia (a disorder with psychological and social components) is partially characterized by widespread musculoskeletal pain with allodynic and hyperalgesic components. While in some respects symptomatically similar to MPS, muscles of fibromyalgia patients do not present with taut bands of hypercontracted myofibers [19]; instead, muscles throughout the body present with tender points [3, 16, 20]. There is some disagreement as to whether the appearance of tender points in muscles and the occurrence of musculoskeletal pain throughout the body are generalized events in patients suffering from fibromyalgia. Some believe that the regionally localized onset and spread of MPS is a characteristic which distinguishes it from the more generalized occurrence of tender points and musculoskeletal pain associated with fibromyalgia [16, 19, 21]. Still other evidence suggests that some forms of fibromyalgia begin with local or regional pain that later precedes widespread musculoskeletal pain [22–25]. Regardless of these points of contention, the underlying mechanisms generating the sensation of musculoskeletal pain associated with fibromyalgia likely significantly overlap with the pathways of pain perception in most muscle injury and disorder models.
Neural Mechanisms of Musculoskeletal Pain Perception
Peripheral Mechanism: The Nociceptive System
Nociceptors are peripheral afferent sensory neurons with free nerve endings located in peripheral tissue, which also make synaptic connections with ascending second-order central sensory neurons in the dorsal horn of the spinal cord. Nociceptors are either thinly myelinated (group III; Aδ-type) fibers or unmyelinated (group IV; C-type) fibers. There is ample evidence from animal studies that both group III and group IV afferent nociceptors are present and active in skeletal muscle of mammals [26, 27]. Nociceptors respond to both noxious mechanical stimuli and chemical stimuli associated with damaged tissue. Importantly, in normal physiologic conditions, nociceptors associated with skeletal muscles are not activated by normal muscle contraction or stretch, or any other non-noxious stimulus and are thus considered high-threshold mechanosensitive. Of note, similar to sensory receptors associated with cutaneous tissue, nociceptors may respond to both chemical and mechanical stimuli (polymodal) or respond only to chemical or mechanical stimuli (unimodal) [28].
Muscle-associated nociceptors are classically activated by a number of humoral and cellular factors (summarized in Fig. 5.1), including the peptide bradykinin (BK) , the modified amine paracrine hormone serotonin (5-HT), the lipid prostanoid prostaglandin E2 (PGE2), and adenosine triphosphate (ATP). Additionally, locally high potassium ion (K+) concentration activates subsets of nociceptors [6, 27, 29–31]. BK is a humoral peptide enzymatically cleaved from a precursor plasma globulin, high molecular weight (HMW) kininogen, which is synthesized in the liver. Enzymatic cleavage of HMW kininogen to form BK is mediated by the plasma protease kallikrein [32]. BK exerts it effects through the kinin B1 and B2 G-protein-coupled receptors. In homeostatic conditions, BK generally binds to the B2 receptor expressed by skeletal muscle-associated nociceptive neurons. However, during inflammatory events, the B1 receptor is upregulated in nociceptive nerve endings, and BK subsequently exerts algesic musculoskeletal effects via this G-protein-coupled receptor which is excitatory to muscle nociceptors upon activation [33–35].
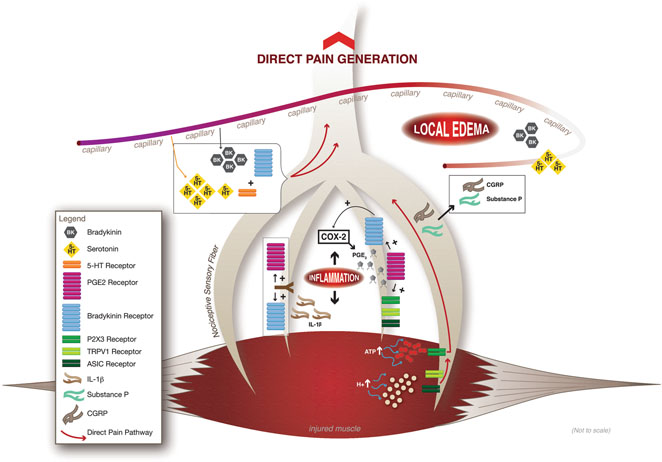
Fig. 5.1
Summary model of the humoral and local origins of musculoskeletal pain. At the onset of inflammation following muscle injury, the humoral protein bradykinin (BK) increasingly binds to the upregulated kinin B2 receptor expressed by nociceptive neurons, activating them. Additionally, upregulated in circulation by muscle inflammation is serotonin (5-HT), which both activates nociceptive neurons and increases their sensitivity to BK. Further, sensitizing nociceptive neurons to BK is the inflammation-activated, locally synthesized lipid prostaglandin E2 (PGE 2 ). Local pain factors arise following nociceptor activation with the release of the peptides substance P (SP) and calcitonin gene-related peptide (CGRP) which serve as vasodilators to induce local edema. With the onset of edema, there is local increase in the production of the cytokine interleukin-1 beta (IL-1β) which increases PGE2 synthesis and the receptors for both PGE2 and BK. Other local factors influencing musculoskeletal pain are the increased concentrations of extracellular adenosine triphosphate (ATP) due to damaged muscle membranes following injury. ATP via activation of the P2X3 receptor can directly activate nociceptive neurons. In addition, with advancing local edema and ischemia associated with muscle damage, the pH of the local cellular environment drops, thus increasing the concentration of protons (H+). The H+-activated transient receptor potential cation channel subfamily V, member 1 (TRPV1) and the acid-sensing ion channels are stimulated by the increased H+ concentration and directly activate muscle-associated nociceptors. (Courtesy of Elizabeth A. Drum)
Studies demonstrate that 5-HT can exert a direct and independent algesic effect when injected into skeletal muscle [36]. Importantly, if 5-HT injection precedes BK injection by only a few minutes, the resulting musculoskeletal pain is significantly increased [37–40]. Thus, 5-HT seems to act both independently as a nociceptive factor and synergistically with BK to hypersensitize nociceptors to increased local BK concentrations following muscle injury . In both human and animal studies, 5-HT injection elicits immediate muscle tenderness, but concurrent with this direct algesic effect is the seemly simultaneous hypersensitization of the muscle nociceptors to BK. Supporting this, BK administration following 5-HT injection in these studies moves skeletal muscle from a state of tenderness to a state of musculoskeletal pain when stimulated by otherwise weak, non-noxious mechanical stimuli, a hallmark of hyperalgesia associated with many muscular disorders and injuries [39–41]. BK injection alone does not elicit the same level of pain, thus supporting the hypothesis that nociceptors must undergo a hypersensitization process to generate the level of musculoskeletal pain associated with muscle damage.
In contrast to the direct effects of 5-HT on the onset of musculoskeletal pain, PGE2 does not have any known primary algesic effects on skeletal muscle, as demonstrated by studies showing that injection of the lipid into muscles does not elicit musculoskeletal pain [36]. The role of PGE2 in the onset of muscle pain seems to be to directly hypersensitize musculoskeletal nociceptors to BK. Supporting this idea, muscle nociceptors with a high threshold for mechanical stimuli for activation dramatically increase their sensitivity and propensity for pain generation following PGE2 injection and subsequent BK treatment [36]. The relationship between PGE2 and BK in the sensitization of muscle-associated nociceptors and the onset of musculoskeletal pain strongly informs our current models and understanding of the discomfort and soreness that results from muscle damage following overload or overuse. Muscle damage leads to local inflammation and increased BK concentrations in the area of insult. Likewise, tissue damage leads to the upregulation and increased activity of cyclooxygenase-2 (COX-2), the enzyme that mediates the conversion of arachidonic acid to active PGE2. The mechanism by which PGE2 hypersensitizes muscle-associated nociceptors to BK is currently unknown. The two PGE2 receptor isoforms known to be expressed by nociceptor nerve endings, EP1 (prostaglandin E receptor 1) and EP4 (prostaglandin E receptor 4), are G-protein-coupled receptors, which activate protein kinase C (PKC) and protein kinase A (PKA) pathways, respectively [42]. PGE2-mediated hypersensitization of muscle-associated nociceptors to BK is unlikely to be immediately at the gene transcription regulatory level due to the time course of sensitization (minutes). Rather, the downstream effects of EP1 and/or EP4 activation likely modify the activity of existing B1 receptors expressed by nociceptive neurons, or possibly their affinity for the BK ligand. Supporting this line of reasoning, other known activators of nociceptors , transient receptor potential vanilloid-1 (TRPV1) channels, the purinergic P2X3 receptor, and the tetrodotoxin-resistant voltage-gated sodium (Na+) channel Nav1.9 (each discussed in detail below) are targets of EP1 and EP4 modification [43–45]. Future studies should delineate the specific mechanism by which PGE2 hypersensitizes nociceptors to BK and whether EP1 and/or EP4 signal transduction cascades target the B1 receptor during muscle inflammation.
While PGE2 influences the strength of BK signaling and subsequent nociceptor activation, BK and local signaling factors stored in the free endings of nociceptors regulate PGE2 release in an area of tissue damage. Substance P (SP) and calcitonin gene-related peptide (CGRP) are neuropeptides expressed by nociceptors, but do not directly elicit pain after injection into muscle. However, activation of nociceptors induces the release of SP and CGRP from nerve endings, which go on to act as strong vasodilators and lead to local edema [46]. The induction of edema leads to further increases in both BK and PGE2 release in the area of inflammation and pain generation increases significantly. As edema and inflammatory processes progress, there is a local increased release of the cytokine interleukin-1 beta (IL-1β). IL-1β signaling has been shown to increase both COX-2 and prostaglandin EP1 and EP4 expression [47, 48] as well as upregulate both kinin B1 and B2 receptor gene expression [49, 50]. Thus, a process that some have described as “a vicious cycle” [34] is induced in which signaling factors regulating pain generation and inflammation and edema also enhance each other’s activity. These cyclic signaling pathways are expected to underlie the induction of musculoskeletal pain, tenderness, and possibly TrP formation associated with muscular disorders such as DOMS, fibromyalgia , and MPS, respectively. Current strategies (see inset, Fig. 5.1) to inhibit the recurrent induction of pain and edema pathways include the wide use of nonsteroidal anti-inflammatory drugs (NSAIDS) that specifically inhibit the activity of COX-1 and COX-2 enzymes, thereby reducing prostaglandin signaling. Another promising drug currently under investigation is fasitibant chloride, a selective kinin B2 receptor antagonist. In animal studies, fasitibant chloride has been shown to inhibit the synergistic activity of BK and IL-1β to increase COX-2 gene expression and PGE2 release [51] and serves to significantly reduce inflammatory events [52].
As mentioned earlier, nociceptors are activated by a number of intra- and extracellular signaling molecules. The metabolically important nucleoside triphosphate ATP can bind to the membrane purinergic receptor P2X3 and induce excitatory Na+ currents in nociceptive neurons [53, 54]. Muscle membrane damage due to overload, overuse, or pathological processes leads to ATP diffusion from the myofiber cytoplasm into the extracellular environment. Subsequent activation of P2X3 receptors by ATP is sufficient to induce excitation in muscle-associated group IV afferent nociceptors which leads to pain generation [55, 56]. This mechanism is highly associated with muscle damage due to overload or overuse, as well as injury occurring from eccentric muscle contraction. Other novel activators of nociceptor are the proton (H+)-sensitive TRPV1 channels and acid-sensing ion channels (ASICs). These channels are activated by increased H+ ion concentration or lowered pH [57]. Importantly, lowered pH in muscle tissue is associated with a number of conditions, including overwork, inflammatory processes, and muscular ischemia. A lowered pH of 6.0 is sufficient to activate both TRPV1 and ASICs and subsequently excite musculoskeletal nociceptors [31]. In animal studies, antagonists to P2X3 receptors, TRPV1 channels, and ASICs were able to reverse hyperalgesia processes associated with DOMS following eccentric muscle contractions indicating the significant role these excitatory ion channels play in musculoskeletal pain generation [58]. Also of importance, profound muscular ischemia is associated with the taut bands of the TrPs found in MPS patients [59]. Likewise, reduced blood flow to skeletal muscle has been described as common symptom among those suffering from fibromyalgia [60]. These findings may indicate that a lowered pH and activation of TRPV1 channels and ASICs expressed by musculoskeletal nociceptors may contribute to pain associated with these disorders.
Central Mechanism: Dorsal Spinal Cord and Brain Stem
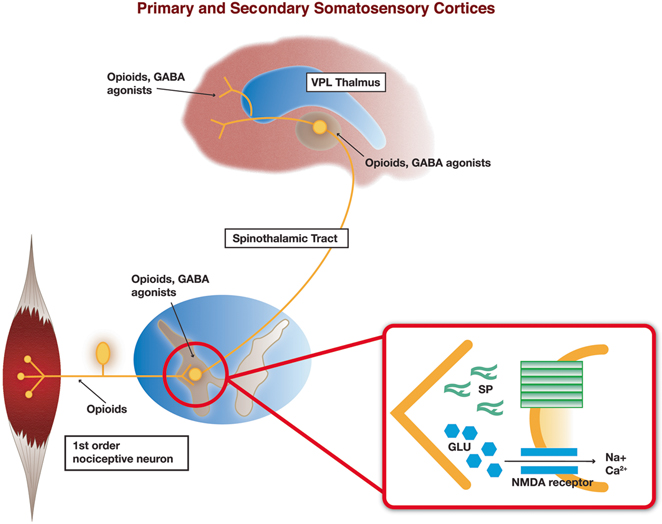
Axonal projections from musculoskeletal nociceptive neurons enter the dorsal root ganglia to provide input to the dorsal horn of the spinal cord and to the trigeminal nucleus pars caudalis of the brain stem (see Fig. 5.2). The sensory dorsal horn of the spinal cord is divided into five laminae, of which laminae III, IV, V, and a small division of lamina II are innervated mainly by hair follicle and tactile sensory Aδ- and Aβ-fibers. The primary nociceptive group III (Aδ-fibers; lightly myelinated) and group IV (C-fibers; unmyelinated) afferents innervating skeletal muscle terminate in dorsal horn lamina I and a large portion of lamina II [61]. Group IV nociceptive neurons are classified as being peptidergic (expressing SP and CGRP) or non-peptidergic [62, 63]. Peptidergic nociceptive group IV afferents include those associated with musculoskeletal tissue, while non-peptidergic afferents generally consist of sensory neurons with nerve endings in the epidermis [64, 65]. The primary neurotransmitter utilized by all nociceptive afferent neurons is the excitatory amino acid glutamate, although SP and CGRP are common neuropeptide co-transmitters [62]. Primary afferent nociceptors make excitatory synapses with second-order neurons in the dorsal horn, which, in turn, ascend in the lateral spinothalamic tract to target the ventral posterior lateral (VPL) and ventral posterior medial (VPM) nuclei of the thalamus. Tertiary sensory neurons of the VPL and VPM carry nociceptive information to the primary and secondary somatosensory cortices [61].