Abstract
Objectives
This review aims to define the concept of neuromuscular fatigue and to present the current knowledge of the central and peripheral factors at the origin of this phenomenon. This review also addresses the literature that focuses on the mechanisms responsible for the adaption to neuromuscular fatigue.
Method
One hundred and eighty-two articles indexed in PubMed (1954–2010) have been considered.
Results
Neuromuscular fatigue has central and peripheral origins. Central fatigue, preponderant during long-duration, low-intensity exercises, may involve a drop in the central command (motor, cortex, motoneurons) elicited by the activity of cerebral neurotransmitters and muscular afferent fibers. Peripheral fatigue, associated with an impairment of the mechanisms from excitation to muscle contraction, may be induced by a perturbation of the calcium ion movements, an accumulation of phosphate, and/or a decrease of the adenosine triphosphate stores. To compensate for the consequent drop in force production, the organism develops several adaptation mechanisms notably implicating motor units.
Conclusion
Fatigue onset is associated with an alteration of the mechanisms involved in force production. Then, the interaction between central and peripheral mechanisms leads to a series of events that ultimately contribute to the observed decrease in force production.
Résumé
Objectifs
Cette revue vise à définir la notion de fatigue neuromusculaire et à présenter les connaissances actuelles relatives aux facteurs centraux et périphériques à l’origine de ce phénomène au niveau du muscle sain. Cette revue aborde également la littérature traitant des mécanismes mobilisés afin de s’adapter à la fatigue.
Méthode
Cent quatre-vingt-deux articles indexés dans PubMed (1954–2010) ont été considérés.
Résultats
La fatigue neuromusculaire a des origines centrales et périphériques. La fatigue centrale, prépondérante pour les exercices de faible intensité et de longue durée, impliquerait une baisse de la commande centrale (cortex moteur, motoneurone) influencée par l’activité des neurotransmetteurs cérébraux et les afférences musculaires. La fatigue périphérique, associée à une altération des mécanismes allant de l’excitation à la contraction musculaire, serait induite par une perturbation des mouvements d’ions calcium, une accumulation de phosphate, et/ou une baisse des réserves en adénosine triphosphate. Pour pallier cette diminution de la force produite, l’organisme développe plusieurs mécanismes adaptatifs impliquant notamment l’activité des unités motrices.
Conclusion
L’apparition de la fatigue est accompagnée d’une altération des mécanismes impliqués dans la production de force. L’interaction entre ces mécanismes centraux et périphériques engendre alors une cascade d’événements participant à la baisse de la force produite.
1
English version
1.1
Definition
In common parlance, “fatigue” is a term used to describe the decrease in physical performance associated with an increase in the real and/or perceived difficulty of a task or exercise . During muscle exercise, fatigue is defined as the inability to maintain the required level of strength . This definition is associated with the notion of a “break point” and the sudden appearance of fatigue and inability to sustain the exercise. However, many neurophysiological mechanisms are perturbed before the body feels the effect of fatigue and these changes sometimes constitute advance warning of fatigue. Furthermore, the initial state of the neuromuscular system (e.g. energy reserves, ion concentrations and the arrangement of contractile proteins) is altered as soon as exercise starts. Fatigue then develops progressively until the muscle is no longer able to perform the requested task. Neuromuscular fatigue, therefore, represents “any exercise-induced reduction in force or power regardless of whether the task can be sustained or not” . Fatigue is a complex, multifactorial phenomenon whose mechanisms are influenced by the characteristics of the task being performed (i.e. type and duration of the exercise, speed and duration of the muscle contraction ). The latter can be modified by the investigator wishing to impose fatigue in a standardized context or by a trainer seeking to change the characteristics of his/her training session, for example.
The physiological processes involved in muscle force generation extend to the whole neuromuscular system. Many different factors may underlie and/or be involved in the expression of neuromuscular fatigue. Moreover, the maintenance of submaximal strength over time results from facilitatory and inhibitory influences of neuromuscular origin. In fact, the neuromuscular system tries to compensate for the decrease in force generation by implementing a variety of nervous and muscle-related mechanisms, in order to delay the point at which the task can no longer be performed.
1.2
Causes of fatigue
During the performance of maximal or submaximal muscle exercise, the decrease in force generation is caused by several different physiological phenomena . Hence, “central fatigue” designates a decrease in voluntary activation of the muscle (i.e. a decrease in the number and discharge rates of the motor units (MUs) recruited at the start of muscle force generation), and “peripheral fatigue” indicates a decrease in the contractile strength of the muscle fibres and changes in the mechanisms underlying the transmission of muscle action potentials . These phenomena occur at the nerve endings and at the neuromuscular junction (NMJ) and are usually associated with peripheral fatigue.
The set of sites which can lead to a decrease in force generation are shown in Fig. 1 . Hence, one can see alterations in:
- •
(1) activation of the primary motor cortex;
- •
(2) propagation of the command from the central nervous system (CNS) to the motoneurons (the pyramidal pathways);
- •
(3) activation of the MUs and muscles;
- •
(4) neuromuscular propagation (including propagation at the NMJ);
- •
(5) excitation-contraction coupling;
- •
(6) availability of metabolic substrates;
- •
(7) state of the intracellular medium;
- •
(8) performance of the contractile apparatus;
- •
(9) blood flow.
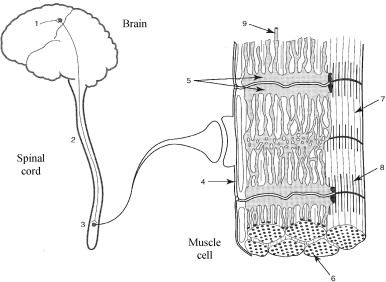
One can distinguish between central sites (1 to 3) and peripheral sites (4 to 9) that underlie fatigue. Furthermore, Table 1 covers the mechanisms and physiological sites that can be affected by neuromuscular fatigue and thus may be the source of a decrease in force generation.
| Central fatigue |
| 1. Propagation of axonal action potentials may be blocked at axonal branching sites, inducing a loss of activation of the muscle fibre. The significance of this factor remains to be determined |
| 2. Motoneuron command may be influenced by reflex activities induced by the muscle afferents. Hence, central fatigue could (to some extent) be compensated for by reflexes due to mechanoreceptors (neuromuscular spindles and Golgi tendon organs) |
| 3. The stimulation of type III and IV nerves (chemoceptive and nociceptive afferents) may induce a drop in the motoneuron discharge rate and an inhibition of motor cortex command |
| 4. The excitability of the cells within the motor cortex may vary during a sustained motor task |
| 5. The synaptic effects of serotoninergic neurons could augment and, thus, induce an increase in the sensation of fatigue. This could occur after an increase in the brain’s uptake of the serotonin precursor tryptophan. During prolonged exercise, this type of increase could be related to the drop in the plasma concentration of branched chain amino acids |
| 6. The exercise could lead to the release of cytokines such as interleukin-6, which is associated with the sensation of fatigue |
| Peripheral fatigue |
| A. Changes in the intracellular environment |
| 1. Accumulation of lactate and hydrogen ions. The accumulation of hydrogen ions is partly buffered by bicarbonate, which induces a release of carbon dioxide. This changes the respiratory quotient |
| 2. Accumulation of ammonia |
| 3. Accumulation of heat, which induces greater sweat secretion. The water loss associated with this phenomenon could lead to dehydration |
| B. Changes within the muscle fibres |
| 1. Accumulation of inorganic phosphate in the sarcoplasma, inducing a drop in the contractile force due to inhibition of the cross-bridges’ interactions |
| 2. Accumulation of hydrogen ions in the sarcoplasma, inducing a drop in the contractile force due to inhibition of the cross-bridges’ interaction. Moreover, this accumulation could trigger impaired reuptake of calcium by the sarcoplasmic reticulum (SR). This could be the main cause of the extended relaxation period after a fatiguing contraction |
| 3. Accumulation of magnesium ions in the sarcoplasma. The magnesium ions may limit calcium release by the SR |
| 4. Inhibition of the calcium release by the SR, due to accumulation of inorganic phosphate. Calcium release is inhibited by (i) precipitation of calcium phosphate and (ii) phosphorylation of the calcium channels |
| 5. A decrease in glycogen reserves and (in extreme cases) a drop in blood glucose. The depletion of glycogen reserves leads to fatigue through a mechanism that is not yet well understood. A drop in blood glucose – even a temporary one – could strongly perturb the operation of the central nervous system |
| 6. Drop in the nerve action potentials’ speed of propagation along the sarcolemma, probably as a result of biochemical changes inside and around the muscle fibres. The fall in the propagation speed is illustrated by a change in the frequency content of the electromyographic signal but has no known immediate effect on force generation |
| 7. Increase in the efflux of potassium ions from the muscle fibres. The increase in potassium in the lumen of the transverse tubules could block action potentials at this point and hence diminish the force generated, due to impaired excitation-contraction coupling |
1.2.1
Central factors in fatigue
Central fatigue can be defined as a progressive, exercise-induced degradation of the muscle voluntary activation . Central fatigue encompasses all the supraspinal and spinal physiological phenomena capable of inducing a decrease in motoneuron excitation.
The presence of central fatigue can be evaluated by using percutaneous electrical stimulation . During maximal voluntary contraction (MVC), the observation of a force peak following an electrical stimulation superimposed on the motor nerve innervating the muscle indicates that the voluntary activation was not in fact maximal. This means that some MUs are either not recruited or do not fire often enough for the muscle fibres to generate maximal force . Over the course of fatiguing exercise, an increase in this superimposed peak force may be associated with progressive impairment of voluntary activation (for a review, see ) and thus with the presence of central fatigue . Hence, Löscher et al. reported the presence of central fatigue during isometric contraction of the foot flexors (30% MVC). Using electrical stimulation, Kent-Braun and Schillings et al. have estimated that 20 and 12% of the loss of strength during a maximal isometric contraction of the ankle dorsiflexors and of the arm flexors are due to central fatigue, respectively. Furthermore, several transcranial magnetic stimulation (TMS) studies by Gandevia’s group have shown that central fatigue can account for over 25% of the drop in force seen during sustained, maximal contractions . However, central fatigue appears to contribute more significantly to the decrease in force generation during low-intensity exercise . In fact, Smith et al. have indicated that two thirds of fatigue can be attributed to central (supraspinal) mechanisms during isometric flexion of the forearm at 5% MVC.
One of the causes of weaker central command during prolonged exercise could be the decreased excitation supplied by the motor cortex . This supraspinal fatigue can be identified by using TMS or transcranial electrical stimulation . In fact, TMS activates motor cortex neurones and thus, induces contraction of the muscles innervated by the latter . To study the development of supraspinal fatigue, the subject is asked to perform short MVCs at regular intervals during sustained, submaximal, isometric exercise. Over the course of the effort, an increase in the amplitude of the TMS-induced peak force during the maximal contraction indicates the presence of supraspinal fatigue, which may be due to submaximal excitation by the motor cortex . Several authors have used TMS to shown a decrease in the excitation supplied by the motor cortex during sustained, isometric arm flexion between 15 and 30% MVC . Furthermore, Soggard et al. estimated that the supraspinal fatigue represented about 40% of the total loss of strength during isometric flexion of the elbow at 15% MVC until exhaustion.
The causes of supraspinal fatigue are poorly known. However, several explanatory hypotheses have been suggested. Firstly, supraspinal fatigue may be linked to the depletion or accumulation of certain brain neurotransmitters, inducing a decrease in corticospinal descending excitation. The most-studied neurotransmitter in this context is serotonin (5-hydroxytryptamine [5-HT]). Newsholme et al. have suggested that prolonged exercise increased the brain’s serotoninergic activity, which in turn limits central command and thus, the recruitment of MUs. This situation may be related to serotonin’s effects on lethargy and loss of motivation .
Serotonin cannot cross the blood-brain barrier. The brain’s neurones must thus synthesize this compound themselves from its precursor, tryptophan (TRP). In the plasma, free TRP competes with branched chain amino acids (BCAAs) for transport into the brain. Thus, the brain’s synthesis of serotonin increases as the plasma free TRP/BCAA ratio rises. This may occur during prolonged exercise, since BCAAs are used by the muscles to produce energy (causing a fall in plasma BCAA levels) and because free fatty acids (the concentration of which increases during exercise) displace albumin-bound TRP (causing an increase in free TRP levels). Several methods have been used to reveal the influence of the brain’s serotoninergic activity on central fatigue and physical performance. Studies on the dietary supply of TRP and BCAAs have given contradictory results concerning the influence of these nutrients on the ability to sustain exercise . In contrast, a supply of carbohydrates limits the activity of TRP and prolongs cycling exercise at maximal aerobic power . The ingestion of carbohydrates may:
- •
reduce lipolysis and thus the plasma concentration of circulating free fatty acids;
- •
counter the exercise-induced increase in free TRP and thus, limit serotonin synthesis in the brain.
This has been confirmed by recent work demonstrating that ingestion of carbohydrates during a 3-hour cycling test at 200 W limited the consumption of brain TRP and prevented hypoglycaemia . Other work on serotonin reuptake and the administration of serotonin agonists or antagonists has confirmed that this neurotransmitter plays a significant role in the initiation and continuation of physical exercise . Central fatigue could also be influenced by the activity of other neurotransmitters. Dopamine may limit serotonin synthesis by acting on TRP hydroxylase, the key enzyme in 5-HT synthesis . More generally, catecholamines (e.g. adrenaline, noradrenaline and dopamine) may have an effect on fatigue by virtue of their effect on motivation and motor action . Furthermore, amphetamine (a dopamine analogue) improves physical performance . It has also been suggested that other neurotransmitters (such as glutamate, acetylcholine, adenosine and gamma-aminobutyric acid [GABA]) are involved in the development of central fatigue . Interactions between these neurotransmitters could also influence central fatigue . However, there are too few published data to draw clear conclusions on the influence of these neurotransmitters on the onset of fatigue. In addition, changes in the concentrations of these various neurotransmitters may also alter cognitive responses or may be involved in subconscious mechanisms of fatigue .
Furthermore, central command may be limited by changes in the brain concentration of other substances, such as ammonium ions and glycogen. During exercise, the plasma concentration of ammonium increases (mainly due to deamination of BCAAs ). Ammonium ions can easily cross the blood-brain barrier and so their accumulation could thus be a factor in the drop in motor cortex activity during prolonged exercise, via its effects on cerebral brain blood flow, the activity of certain neurotransmitters and synaptic transmission . Glycogen could also play a role in central fatigue because brain activation is associated with a drop in brain glycogen . In fact, the brain has low glycogen reserves and the latter are rapidly exhausted. Although these reserves can be rapidly renewed, depletion could influence brain function and, notably, may have an effect on the activity of serotonin . The influence of the brain’s neurotransmitter, ammonium and glycogen levels on central fatigue and performance has been evidenced in prolonged exercise (over 30 minutes) . Likewise, hyperthermia (a phenomenon sometimes associated with sports exercise) may reduce the activity of central nervous command . Lastly, a new hypothesis has emerged with respect to the involvement of humoral signals (cytokines) produced by muscle activity in the presence of fatigue . Nevertheless, the mechanisms linking cytokines to fatigue are still poorly understood and require further research.
Another hypothesis concerning supraspinal fatigue suggests that certain muscle afferents related to the muscles’ biochemical status and force generation capacity limit cortical activity. By looking at electrostimulation-induced fatigue of the interosseous muscles, Pitcher and Miles demonstrated that supraspinal fatigue (as evidenced by a decrease in the motor-evoked potentials) could be modified by muscle afferents. However, the latter authors did not comment on whether this supraspinal fatigue occurred in the motor cortex or further upstream. In an comprehensive survey of the literature, Gandevia gathered evidence of supraspinal inhibition. This author stated that feedback on the muscles’ biochemical status and force generation capacity is likely to reduce stimulation from cortical sites. The role of muscle afferents and the association between peripheral fatigue and supraspinal fatigue can be demonstrated by using ischaemia to prevent recovery from muscle fatigue at the end of a fatiguing contraction . At the end of the voluntary contraction, the fatigue-induced modifications in nervous command can be “recovered”, whereas ischaemia prompts the group III and IV muscle afferents (which are sensitive to metabolites produced during fatigue) to continue to discharge. Measurement of voluntary activation by TMS then showed that as long as the muscle remained ischaemic, the supraspinal fatigue persisted . In contrast, corticospinal and motoneuron activities appear to return to normal values . This suggests that a mechanism related to the muscle’s fatigue status acts to limit voluntary activation but does not act on the motoneurons or motor cortex excitation. It is, therefore, possible that the fatigue-sensitive muscle afferents limit voluntary command by acting upstream of the motor cortex , as suggested by Pitcher and Miles .
At the spinal level, central fatigue may be caused by several mechanisms. Firstly, the inhibitory afferents from intramuscular receptors appear to be involved in the decrease in motoneuron activity. In fact, Bigland-Ritchie et al. have shown that the motoneurons’ discharge rate can be regulated by peripheral reflexes in response to fatigue-induced metabolic variations within the muscle. Hence, group III and IV muscle afferents (metaboreceptors) appear to be stimulated by ischaemia , hypoxaemia and the extracellular accumulation of potassium (K + ) and lactate . Stimulation of these metaboreceptors during fatiguing exercise may then inhibit the activity of the alpha motoneurons . The neuromuscular spindles (group Ia and II afferents) are oriented parallel to the muscle fibres and provide the nervous system with information on the muscle’s length and changes in length . Macefield et al. reported that the discharge rate of these receptors decreased progressively during an isometric contraction below 30% MVC. This decrease was intense at the start of exercise but then changed more slowly. The drop in discharge rate induced by neuromuscular spindles may thus be associated with the limitation of alpha motoneuron activity . Furthermore, the sensitivity of these neuromuscular spindles can be reduced by structural changes in muscle (e.g. variations in musculotendinous stiffness), which in turn can be induced by repeated contraction or stretching . Changes in muscle stiffness could also influence the muscle’s electrical activity and contribute to fatigue . Nevertheless, how these changes in stiffness might influence the muscle’s contractile activity remains to be determined.
The Golgi tendon organs (group Ib afferents) at the musculotendinous and musculoaponeurotic junctions provide the CNS with feedback on the intramuscular tension . These mechanoreceptors are thought to inhibit neuronal activity . The Golgi tendon organs’ exact effects on motoneuron activity during a fatiguing task are difficult to determine because these afferents are difficult to isolate and their projections include interneurons receiving signals from Ia afferents . However, the influence of the signals emitted by the neuromuscular spindles and the Golgi tendon organs has hardly been studied and is poorly characterized – mainly because their effects can be attenuated at the spinal level and is less intense than those of many group III and IV afferents .
In addition, the decrease in motoneuron activity could also be due to the latter’s intrinsic properties . According to the latter authors, the motoneuron may adapt its discharge rate during fatigue in response to constant excitation. The stimulation of group III and IV afferents could induce changes in the motoneuron membrane’s intrinsic properties, modifying the cell’s discharge rate . This decrease in the discharge rate (late adaptation) may, therefore, be more marked for fast-twitch fibres than for slow-twitch fibres . However, data on this phenomenon are scarce and have only been gathered in animal experiments.
Furthermore, intracortical inhibition could also be involved in the drop in muscle performance under fatiguing conditions. Recently, McNeil et al. suggested that during a 2-minute MVC of the elbow flexors, intracortical inhibition increased as the fatigue progressed.
Lastly, motoneurons (mainly those in fast-twitch MUs) are inhibited by Renshaw cells, which are stimulated by these same motoneurons and by descending and peripheral influences. By using the indirect Hoffmann reflex method (a muscle response induced by excitation of group Ia afferents during electrical stimulation), several studies have suggested that this inhibition increases during maximal efforts but falls during submaximal contractions at 20% of the MVC, when central fatigue occurs .
Hence, many anatomic sites and physiological mechanisms are affected by central neuromuscular fatigue. This fatigue also appears to perturb normal processes in the peripheral part of the neuromuscular system.
1.2.2
Peripheral factors in fatigue
The factors involved in peripheral fatigue include alterations in neuromuscular transmission, muscle action potential propagation, excitation-contraction coupling and related contractile mechanisms ( Fig. 1 ).
Neuromuscular transmission is defined as transformation of the nerve action potential (AP) into a muscle action potential and takes place at the neuromuscular junction. During fatigue, this mechanism can be altered by:
- •
insufficient propagation of the nerve potential at the nerve endings;
- •
a failure of the coupling between excitation and neurotransmitter secretion in the synaptic gap;
- •
neurotransmitter depletion;
- •
reduced neurotransmitter release;
- •
a decrease in the sensitivity of the post-synaptic acetylcholine receptors and the post-synaptic membrane .
During sustained contraction, fatigue may decrease the excitability of small-diameter axons. A slight decrease in excitation may then lead to inactivation of these axons and a decrease in the amount of neurotransmitter released in the synaptic gap . These authors have thus reported an in vitro study in which certain nerve endings of the motoneurons innerving the rat diaphragm were no longer activated after several minutes of electrical muscle stimulation. This phenomenon was not dependent on the K + or calcium ion (Ca 2+ ) concentrations or pH in the physiological saline bath. The authors thus concluded that the intramuscular oxygen concentration had an influence on propagation of the AP. Moreover, the amplitude of motor end-plate potentials decreases during fatiguing exercise, as a result of a decrease in the quantity of neurotransmitter (acetylcholine) released by each nerve ending. This may be related to a reduction in the number of exocytotic vesicles and/or a decrease in the quantity of acetylcholine released per vesicle . Furthermore, work by Van Lunteren and Moyer has suggested that variations in the intracellular Ca 2+ concentration within the nerve endings play a key role (via exocytosis) in the quantity of neurotransmitter released. However, this work was performed in vitro in the animal, which limits the conclusions that can be drawn about the involvement of the said phenomena in voluntary movements in humans. The efficiency of neuromuscular transmission is also dependent on the ACh-sensitivity of the postsynaptic membrane receptors. In fact, prolonged exposure of cholinergic receptors to ACh appears to desensitize them to this neurotransmitter and translates into a lengthening of the time required for ACh to bind to its receptor . However, the relevance of postsynaptic receptor desensitization within the fatigue process (just like the other mechanisms that can degrade neuromuscular transmission) has not been established with certainty. The impairment in neuromuscular transmission can be illustrated by applying electrical M waves before, during and after a fatiguing contraction. The M wave corresponds to the muscle’s electrical activity (measured by electromyography) in response to electrical stimulation of the nerve . A decrease in the M wave’s amplitude can then be interpreted as degradation of one or several pathways involved in the conversation of the axonal action potential into the muscle action potential (MAP). Propagation of the signal over the sarcolemma has a considerable influence on the amplitude of the M wave. However, the results obtained using this electrical stimulation technique are subject to debate in the literature and doubt has been cast on their relevance under some circumstances . In fact, the M wave’s characteristics can be affected by factors such as the synchronisation of the muscle fibres’ action potentials and/or the extent of neurotransmitter dispersion during its release . Furthermore, the changes of the M wave may not reflect variations in intracellular action potentials . In this respect, some authors have highlighted the advantages of high-density EMG which, by using electrode arrays, adds spatial information to the temporal information contained in the EMG signal .
The results regarding the effects of fatigue on changes in signal propagation are inconsistent . Hence, some studies report that the impairment of neuromuscular transmission appears to be a factor that limits muscle excitation and contributes to the drop in force generation, which seems to occur more during low-intensity, long-duration exercise than intense, short-duration contractions . Nevertheless, other researchers have not observed any change in the M wave following submaximal contractions lasting less than 5 minutes . It appears thus difficult to draw firm conclusions on the influence of neuromuscular transmission on force generation under fatiguing conditions.
Muscle action potentials are generated when the sum of the excitatory and inhibitory presynaptic potentials reaches or exceeds the muscle cells’ excitability threshold. However, several factors are likely to alter the transformation of the nervous excitation into muscle force and fatiguing exercise may contribute to this phenomenon : the propagation of the MAP along the sarcolemma and the transverse tubules, the sarcoplasmic reticulum’s (SR) permeability to Ca 2+ , the movement of Ca 2+ inside the sarcoplasma, the active efflux of Ca 2+ from the SR, Ca 2+ binding to troponin and the myosin-actin interaction associated with the work performed by the cross-bridges. Calcium plays an essential role in the mechanisms which lead to force generation. Westerblad and Allen observed that during prolonged isometric contraction, the developed force started to decrease even when the intramuscular Ca 2+ concentration was high – suggesting degradation of force generation at the myofibrillar level. In a second phase, the decrease in force was associated with a drop in the amount of Ca 2+ released by the SR.
Of the many metabolic changes associated with prolonged muscle contraction, two in particular appear to underlie the myofibril’s reduced force generation capacity during fatigue : the increase in intracellular concentrations of hydrogen ions (H + ) and inorganic phosphate (Pi). The Pi concentration increases during exercise, due to:
- •
the dissociation of phosphocreatine into Pi and creatine;
- •
the hydrolysis of adenosine triphosphate (ATP).
Hydrogen ions accumulate as a result of the hydrolysis of ATP and the production of lactic acid (which occurs when glycolytic activity exceeds the mitochondria’s oxidative capacity or when the oxygen supply is limited). The accumulation of H + has long been considered to be a significant cause of the decrease in myofibrillar force generation, since it is associated with a decrease in intracellular pH, which perturbs chemical reactions. Nevertheless, these results were obtained at sub-physiological temperatures. In fact, some authors have shown that the acidification of the medium had a very limited effect on the force generation capacities at normal body temperatures . If the acidosis is not the cause of the drop in force generation, then the accumulation of Pi must be. It has been established at low temperatures (10°–15°) that an intracellular increase in Pi limits force generation when the medium is saturated with Ca 2+ . However, work testing temperature variations yielded contradictory results with respect to the influence of Pi on the force generated . Since then, Pi’s influence on force generation mechanisms has been demonstrated experimentally by reducing the concentration of this ion in the medium, which induces an increase in force . Inorganic phosphate could perturb force generation by decreasing the myofibrils’ sensitivity to Ca 2+ and by acting directly on the cross-bridges . In fact, the increase in the Pi concentration under fatiguing conditions may perturb the cross-bridges’ contraction-relaxation cycles . Taken as a whole, these results suggest that the decrease in force generation observed at the very start of contraction (when the intracellular Ca 2+ concentration is high) may be due to the accumulation of Pi.
Next, when muscle work is sustained, the drop in force generation may be related to a decrease in the quantity of Ca 2+ released by the SR. This phenomenon may occur later in fibrils with strong oxidative capacities and sooner in fatigued fibres under anaerobic conditions . Variations in Pi and ATP concentrations appear to be responsible for the impaired release of Ca 2+ . An exercise-induced accumulation of Pi within the cell muscle may favour penetration into the SR. The Pi may then bind to Ca 2+ to yield calcium phosphate (Ca 2+ -Pi), which would thus limit the reserves of Ca 2+ capable of being released from the SR’s cisternae into the sarcoplasma .
Furthermore, the quantity of Ca 2+ released from the SR appears to be related to the ATP concentration. In fact, four sites involved in Ca 2+ release are sensitive to ATP levels . Firstly, Ca 2+ release depends on propagation of the sarcolemma’s muscle action potential to the transverse tubules and on to the SR. During prolonged contraction, a train of action potentials induces a disequilibrium between the Na + and K + concentrations on either side of the cell membrane. The ATP-driven Na + /K + pumps use the energy supplied by the hydrolysis of ATP to move three Na + ions out of the cell and let two K + ions into the cell in each cycle, in order to restore the cell’s homeostasis. A change in the activity of these pumps (e.g. a drop in the ATP concentration) may perturb the excitability of the membrane and thus, alter the propagation of the AP to the SR and, accordingly, the release of Ca 2+ . Next, the opening of the sarcolemma’s K + channels is related to the intracellular ATP concentration . When these channels open (due to an exercise-induced drop in the ATP concentration), K + ions flood out of the muscle cell. As described above, this ionic movement could alter the AP and limit Ca 2+ release. However, these channels do not appear to act significantly during fatigue . During muscle contraction, Ca 2+ is released by the SR so that it can bind to troponin and enable muscle contraction. Next, Ca 2+ reuptake into the SR occurs. Hence, a decrease in the activity of the ATP-driven pumps performing reuptake of Ca 2+ into the SR might lower the amount of Ca 2+ available for release. However, the pumps’ activity does not appear to impair Ca 2+ release . Lastly, signal transmission between the transverse tubules and the SR and Ca 2+ release are also dependent on ATP reserves. Fatigue and the drop in ATP reserves may induce less phosphorylation of certain sites involved in propagation of the AP and could thus limit Ca 2+ (notably through the SR’s Ca 2+ channels ).
Magnesium ions (Mg 2+ ) may also be involved in the impaired Ca 2+ release because they also participate in operation of the SR. In fact, during muscle activation, an increase in the Mg 2+ concentration in the sarcoplasma reduces Ca 2+ release by the SR . Furthermore, Westerblad and Allen have shown that during exercise, the intracellular concentration of Mg 2+ increases and induces a drop in muscle force generation. This exercise-related increase in Mg 2+ levels may be partly due to the fact that Mg 2+ ions bind to ATP molecules (which are broken down during exercise). Activation of the SR’s Ca 2+ channels also releases Mg 2+ ions and may contribute to the concentration increase .
Lastly, the decrease in Ca 2+ release by the SR may also have other causes . It has been reported that mitochondria in skeletal muscle contain more Ca 2+ after exhausting exercise than those in control muscle . Hence, Ca 2+ capture by mitochondria could be involved in the drop in available Ca 2+ . Some research has also suggested that the muscle fatigue caused by eccentric exercise may induce mechanical rupture between the t-tubules and the SR, leading to impaired excitation-contraction coupling . This dysregulation would lead to inadequate Ca 2+ release from the SR. This type of excitation-contraction uncoupling could explain fatigue during low-frequency, endurance exercise .
This body of work as a whole indicates that the drop in force generation during sustained exercise is mainly related to a drop in the quantity of Ca 2+ released by the SR. This may be primarily due to the intracellular accumulation of Pi and the decrease in ATP reserves associated with fatiguing exercise.
During sustained exercise, the blood supply and reserves of metabolic substrates are also likely to influence performance . During muscle contraction, a reduction in blood flow was one of the first mechanisms to have been identified as being involved in fatigue. During physical activity, an increase in blood flow is necessary to supply the active muscles with substrates, evacuate metabolites and dissipate heat. However, muscle contraction often compresses the blood vessels and thus decreases the blood supply to the active muscles; indeed, isometric contraction may even trigger complete ischaemia. If the contraction continues, limitation of the blood supply limit will reduce the oxygen supply and promote the activity of anaerobic metabolic pathways. The drop in blood flow will trigger more rapid accumulation of the metabolites associated with muscle contraction (e.g. Pi, H + ) and will thus accelerate the process of fatigue and the drop in force generation. Sjogaard et al. have shown that the blood supply decreases as exercise intensity increases during leg extensions between 5 and 50% MVC. Furthermore, the greater the contraction intensity, the earlier ischaemia will occur . In this respect, Crenshaw et al. have shown that the maintenance time of leg extensor exercise at 25% MVC coincided with the time to complete ischaemia . Nevertheless, the blood supply is probably not changed by contractions performed at less than 15% MVC .
In most muscle contractions, metabolic substrate availability does not constitute a performance-limiting factor. However, during certain types of endurance exercise (such as cycling at submaximal intensity, i.e. 70 to 80% of the maximal aerobic power), the inability to maintain the required force appears to coincide with the depletion of the leg extensors’ glycogen reserves . Furthermore, intake of additional glucose enables the subjects to maintain the exercise for longer .
However, some authors suggest that failure to sustain high-intensity aerobic exercise is due to the subject’s lack of commitment and perceived effort, rather than exercise-induced central and/or peripheral fatigue . The psychobiological model has nevertheless yet to be validated.
1.3
Adaptations to fatigue
Neuromuscular fatigue (both central and peripheral) will cause the neuromuscular system to adapt to effort in a number of ways, in order to sustain force generation ( Fig. 2 ). The following section will deal with the main adaption mechanisms involved when neuromuscular fatigue occurs during short- to medium-duration maximal and submaximal exercise. There are three types of mechanisms:
- •
modulation of MU activity;
- •
increases in the efficiency of muscle activity (i.e. potentiation);
- •
the “protective” mechanism called “muscle wisdom”.
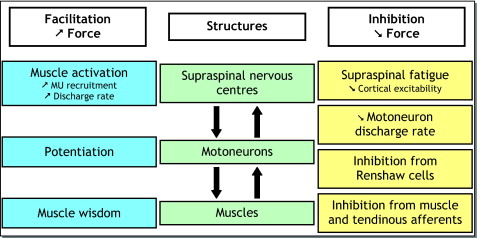
The specific mechanisms associated with very long-term fatigue are not addressed.
1.3.1
Modulation of MU activity
At the muscle level, the main mechanism for dealing with fatigue during submaximal exercise is a combination of increased MU recruitment and modulation of the MU discharge rate. The MU discharge rate appears to fall during sustained, maximal contractions . In contrast, the results relating to submaximal exercise are disparate; fatigue may induce an increase in the MU discharge rate , a decrease or no significant variation, depending on the study and task in question . In any case, the changes in the discharge rate remain small. The observed divergences may be explained by the variety of protocols used, notably in terms of the sustained, submaximal force generated. Kuchinad et al. studied MU discharge rates during low force generation (25% MVC) and moderate force generation (42–66% MVC). The MU discharge rate increased for low-intensity exercise and decreased for higher intensity exercises. These results may reflect differing levels of reflex inhibition in the two exercises. The inter-subject variability in voluntary command during fatigue also appears to be involved in changes in the MU discharge rate . Once an MU is active for a certain duration (depending on its characteristics and the time at which it is recruited) during contraction, a decrease in the discharge rate always appears . The discharge rate of newly recruited MUs during exercise is also subject to debate. Hence, some authors have reported that the discharge rate of these MUs remains constant or increases slightly, whereas others indicate that that they behave in the same way as MUs which have been active since the beginning of the contraction (i.e. a drop in the rate). Carpentier et al. have observed that the discharge rate of newly recruited (type II) MUs may increase and then may decrease.
Motor unit recruitment during dynamic contractions has barely been studied. This can probably be explained by the methodological problems inherent to this type of contraction (such as movement of the electrodes on the skin and changes in muscle length and force). Nevertheless, Ivanova et al. reported a drop in the discharge rate for MUs in the biceps brachii, when fatigue was induced by the performance of submaximal flexions-extensions of the forearms. In contrast, other more recent work suggests that the MU discharge rate does not change during dynamic contraction – notably for the triceps brachii . Hence, Griffin et al. reported that the MU discharge rate remained stable during repeated dynamic contractions of the triceps brachii but dropped during an isometric protocol reproducing the same force variations. Maintenance of the MU discharge rate during dynamic contractions could be due to:
- •
greater activation of the neuromuscular spindles;
- •
higher blood flow.
1.3.2
Potentiation
Potentiation can be defined as an increase in the efficiency of muscle activity produced by several mechanisms (increased cortical excitability, the generation of additional stimuli and post-activation ). This phenomenon appears to occur after a brief period of muscle activity and may induce an increase in the electrical and mechanical responses enabling augmentation of the force generated .
At the supraspinal level, the amplitude of the motor-evoked potentials (MEPs) triggered by TMS is a measure of the excitability of the cortical neurones, whereas the silent period following these MEPs provides information on intracortical inhibition . By using this technique, several authors have shown that cortical excitability may increase at the start of a sustained contraction or after the performance of submaximal contractions . However, cortical inhibition also develops during fatigue but later on in the process . Hence, it appears that physical activity changes the excitability of the cortical neurones; this may result from altered excitatory and inhibitory inputs from other cortical areas (e.g. the premotor area and the parietal cortex) or from subcortical structures (e.g. the thalamus and the cerebellum) .
At the spinal level, the performance of repeated or continuous voluntary contractions induces the generation of new nervous stimuli, which are superimposed on the trains of action potentials . These additional stimuli occur less than 20 ms after stimulation and may increase the force generated and decrease muscle fatigability . An increase in Ca 2+ concentration and greater muscle stiffness may underlie this phenomenon . In fact, these stimuli mean that more cross-bridges are formed within the muscle (by virtue of greater Ca 2+ release) and thus, the force generation increases. The presence of these double discharges (doublets) increases with fatigue and occur particularly towards the end of the contraction . Furthermore, the latter authors suggest that the repetition of doublets occurs for loads that induce a MU discharge rate close to the minimum and may thus be more frequent during sustained, low-intensity contractions. Nevertheless, the impact of these doublets on force generation varies from one individual to another .
At the muscle level, another example of potentiation concerns post-activation. In fact, some researchers have found that the performance of a voluntary muscle contraction enables greater force generation in the subsequent contraction . The force gain due to this phenomenon is extremely variable and depends on the muscle in question. For example, stimulation of the extensor digitorum in the rat inducing a 36% drop in force generation was associated with post-activation (and thus a gain in force) in half the muscle fibres . Post-activation may involve several mechanisms, including the kinetics of Ca 2+ release by the SR , the phosphorylation of myosin light chains and the cross-bridges’ force-speed characteristics . It seems that repeated or sustained muscle contraction induces a number of electrical and mechanical mechanisms in the very first instants of the contraction; these changes enable the muscle to develop more force or to maintain the level of force required. These mechanisms may enable the CNS to limit the initial recruitment of additional MUs.
1.3.3
Muscle wisdom
Neuromuscular fatigue can also be considered as an adaption mechanism for limiting muscle activity before the latter becomes too harmful for the body. Hence, Marsden et al. developed the hypothesis of “muscle wisdom”. This phenomenon corresponds to a decrease in the MU discharge rate and a slowing of the muscle contraction speed during fatigue. Muscle wisdom may be a defence mechanism for limiting fatigue by minimizing the drop in membrane excitation and thus Ca 2+ release . It is possible that the muscle limits central excitation via peripheral afferents, in order to feed back information on the decrease in force generation by active myofibrils . The fall in the MU discharge rate may then better match the relaxation speed. Even though the muscle wisdom hypothesis appears to be appropriate for some muscles during maximal contractions, its application is task- and muscle-dependent and may be less valid during sustained, submaximal isometric contractions and dynamic contractions . Since then, Noakes et al. (e.g. ) have mentioned a “central governor” model, in which the brain regulates muscle performance via MU recruitment . The action of this model may be guided by the many receptors and afferents that feed back to the CNS on cell homeostasis, with the goal of preventing the harmful effects of exhausting exercise. However, this model has been criticised and some authors have highlighted its limitations, as revealed by contradictory results . The latter authors indicate that Noakes et al.’s model cannot be applied to all tasks and that muscle performance is not solely limited by MU recruitment.
1.4
Conclusion
Neuromuscular fatigue can be defined as any exercise-induced decrease in a muscle’s ability to develop force or power . Central fatigue involves impairment of voluntary muscle activation and seems to occur particularly during submaximal, low-intensity muscle contractions . It can be due to a decrease in the excitation supplied by the motor cortex and/or a decrease in motoneuron activity . The activities of the brain’s neurotransmitters and muscle afferents appears to be prime factors in the occurrence of central fatigue, which occurs particularly when the triggering exercise is low-intensity . Peripheral fatigue corresponds to an alteration in muscle contraction. It can be induced by perturbations of:
- •
neuromuscular transmission;
- •
the propagation of the muscle action potential;
- •
excitation-contraction coupling;
- •
contractile mechanisms .
Calcium ions are involved in many steps of the muscle contraction. Release of the ions may be limited by an accumulation of inorganic phosphate and/or by a fall in adenosine triphosphate reserves. The acidity of the medium does not appear to limit force generation at physiological temperatures.
During prolonged muscle contraction, the body has several mechanisms for maintaining the level of force produced and resisting fatigue. Hence, new motor units will be recruited to compensate for the fatigue of those activated at the start of the contraction. A sustained contraction also triggers variations in the motor unit discharge rate. The frequency may fall for high-intensity exercise and may increase for low-intensity exercise. Fatigue can also be considered as a mechanism for limiting the harmful effects of exhausting muscle exercise. The muscle afferents provide the nervous system with information on the state of the muscles and appear to be involved in the regulation of fatiguing exercise.
Lastly, the appearance of neuromuscular fatigue is associated with changes in several of these mechanisms, whether central or peripheral. Interactions between these mechanisms lead to a cascade of events which accelerate or decrease the muscle’s force generation capacity.
Conflicts of interest statement
There is no conflict of interest.
2
Version française
2.1
Définition
Communément, la « fatigue » est un terme employé pour décrire une baisse de la performance physique associée à une augmentation de la pénibilité de l’exercice ou de la tâche, qui peut être réelle et/ou perçue par l’individu . Lors d’un exercice musculaire, la fatigue est définie comme l’incapacité à maintenir un niveau de force exigé . Cette définition est associée à la notion de « point de rupture » et à une manifestation soudaine de la fatigue par l’arrêt de l’exercice. Or, de nombreux mécanismes neurophysiologiques sont perturbés avant le constat mécanique de l’effet de la fatigue et la modification de leur fonctionnement constitue parfois un signe annonciateur d’une fatigue. D’ailleurs, l’état initial du système neuromusculaire (e.g. réserves énergétiques, concentrations ioniques, agencement des protéines contractiles) est altéré dès le début de l’exercice, puis la fatigue se développe progressivement jusqu’à ce que le muscle ne parvienne plus à réaliser la tâche demandée. La fatigue neuromusculaire représente alors « toute diminution, induite par l’exercice, de la capacité du muscle à développer une force ou une puissance, que la tâche puisse être maintenue ou non » . La fatigue est un phénomène complexe et multifactoriel dont les mécanismes sont influencés par les caractéristiques de la tâche réalisée (i.e. la durée de l’exercice, le type, la vitesse et le temps de maintien de la contraction ). Celles-ci peuvent être modifiées par l’expérimentateur voulant imposer une fatigue dans un contexte standardisé ou par un entraîneur agissant sur les caractéristiques de sa séance d’entraînement, par exemple.
Les processus physiologiques impliqués dans la production de force musculaire s’étendent à l’ensemble du système neuromusculaire. De nombreux facteurs peuvent être à l’origine et/ou participer à l’expression de la fatigue neuromusculaire. De plus, le maintien d’une force sous-maximale au cours du temps est le résultat d’influences facilitatrices et inhibitrices d’origine neuromusculaire. En effet, le système neuromusculaire essaie de compenser la baisse de la production de force par la mise en place de différents mécanismes nerveux et musculaires afin de repousser l’instant où la tâche ne pourra plus être réalisée.
2.2
Origines de la fatigue
Lors de la réalisation d’un exercice musculaire maximal ou sous-maximal, la dégradation des capacités de production de force est due à différents phénomènes physiologiques . Ainsi, la « fatigue centrale » (ou d’origine centrale) désigne une baisse de l’activation volontaire (i.e. nombre et fréquence de décharges des unités motrices [UM] recrutées à l’origine de la production d’une force musculaire) du muscle et la « fatigue périphérique » (ou d’origine périphérique) une baisse de la force contractile des fibres musculaires et des mécanismes de transmission des potentiels d’action musculaire . Les phénomènes se produisant au niveau des terminaisons axonales et de la jonction neuromusculaire (JNM) sont généralement associés à la fatigue périphérique.
L’ensemble des sites pouvant être à l’origine de la baisse de production de force sont représentés en Fig. 1 . Ainsi, la dégradation peut survenir au niveau :
- •
(1) de l’activation de l’aire primaire du cortex moteur ;
- •
(2) du cheminement de la commande du système nerveux central (SNC) vers les motoneurones (voies pyramidales) ;
- •
(3) de l’activation des UM et des muscles ;
- •
(4) de la propagation neuromusculaire (incluant la JNM) ;
- •
(5) du couplage excitation-contraction ;
- •
(6) de la disponibilité des substrats métaboliques ;
- •
(7) du milieu intracellulaire ;
- •
(8) de l’appareil contractile ;
- •
(9) du flux sanguin.
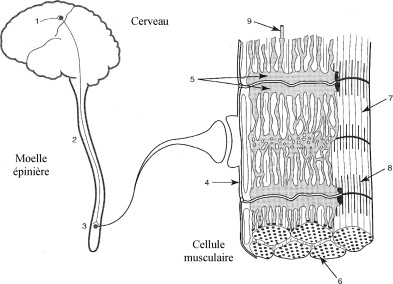
Parmi tous ces sites, on peut distinguer les sites centraux (1 à 3) et les sites périphériques (4 à 9) à l’origine de la fatigue. Par ailleurs, le Tableau 1 regroupe les mécanismes et les sites physiologiques pouvant être affectés par la fatigue neuromusculaire et donc potentiellement à l’origine de la dégradation de la production de force.
| Fatigue centrale |
| 1. La conduction des potentiels d’action axonaux pourrait se bloquer au niveau des sites de liaison, induisant une perte d’activation de la fibre musculaire. L’importance de ce facteur reste indéterminée |
| 2. La commande motrice neuronale pourrait être influencée par des activités réflexes induites par les afférences musculaires. Ainsi, la fatigue centrale pourrait, jusqu’à un certain degré, être compensée par des réflexes issus des mécanorécepteurs (fuseaux neuromusculaires et organes tendineux de Golgi) |
| 3. La stimulation des nerfs de type III et IV (afférences chémoceptives et nociceptives) induirait une baisse de la fréquence de décharge des motoneurones et une inhibition de la commande du cortex moteur |
| 4. L’excitabilité des cellules au sein du cortex moteur pourrait varier au cours d’une tâche motrice maintenue |
| 5. Les effets synaptiques des neurones sérotoninergiques pourrait augmenter, induisant une augmentation de la sensation de fatigue. Cela pourrait survenir suite à une augmentation de l’entrée dans le cerveau du précurseur de la sérotonine, le tryptophane. Lors de l’exercice prolongé, une telle augmentation pourrait être liée à la baisse de la concentration plasmatique en acides aminés à chaîne branchée |
| 6. L’exercice pourrait entraîner la libération de cytokines, comme l’interleukine-6 qui est associée à la sensation de fatigue |
| Fatigue périphérique |
| A. Modifications de l’environnement interne |
| 1. Accumulation de lactates et d’ions hydrogène. L’accumulation des ions hydrogène est en partie tamponnée par le bicarbonate, ce qui induit une libération de dioxyde de carbone. Il en résulte un changement du quotient respiratoire |
| 2. Accumulation d’ammoniaque |
| 3. Accumulation de chaleur qui induit une plus grande sécrétion de sueur. La perte hydrique associée à ce phénomène pourrait mener à la déshydratation |
| B. Modifications au sein des fibres musculaires |
| 1. Accumulation de phosphate inorganique dans le sarcoplasme induisant une baisse de la force contractile due à une inhibition des interactions des ponts d’union |
| 2. Accumulation d’ions hydrogène dans le sarcoplasme induisant une baisse de la force contractile due à une inhibition de l’interaction des ponts d’union. De plus, cette accumulation pourrait provoquer une dégradation du recaptage du calcium par le réticulum sarcoplasmique (RS). Cela pourrait être la cause principale de l’allongement de la période de relaxation après une contraction fatigante |
| 3. Accumulation d’ions magnésium dans le sarcoplasme. Les ions magnésium limiteraient la libération de calcium par le RS |
| 4. Inhibition de la libération de calcium par le RS en raison d’une accumulation de phosphate inorganique. La libération de calcium est inhibée par la précipitation de calcium-phosphate et par la phosphorylation des canaux calciques |
| 5. Baisse des réserves de glycogène et (dans les cas extrêmes) baisse du glucose sanguin. La déplétion des réserves de glycogène mène, d’une façon pas encore parfaitement connue, à la fatigue. Une baisse du glucose sanguin, même temporaire, pourrait fortement perturber le fonctionnement du système nerveux central |
| 6. Baisse de la vitesse de conduction des potentiels d’action nerveux le long du sarcolemme, probablement suite à des changements biochimiques à l’intérieur et autour des fibres musculaires. La baisse de la vitesse de conduction est illustrée par un changement du contenu fréquentiel du signal électromyographique mais n’a pas d’effet immédiat connu sur la production de force |
| 7. Augmentation de la sortie d’ions potassium des fibres musculaires. L’augmentation de potassium dans la lumière des tubules transverses pourrait bloquer les potentiels d’actions au niveau tubulaire et, ainsi, diminuer la force produite en raison d’une dégradation du couplage excitation-contraction |
2.2.1
Facteurs centraux de la fatigue
La fatigue centrale peut se définir comme une dégradation progressive, induite par l’exercice, de l’activation volontaire du muscle . Les facteurs centraux de la fatigue regroupent l’ensemble des phénomènes physiologiques d’origine supraspinale et spinale pouvant induire une diminution de l’excitation des motoneurones.
La présence de fatigue centrale peut être évaluée à l’aide de la stimulation électrique percutanée . Lors d’une contraction maximale volontaire (MVC), l’observation d’un pic de force suite à une stimulation électrique surimposée du nerf moteur innervant le muscle concerné indique que l’activation volontaire n’est pas maximale. Cela signifie que certaines UM ne sont pas recrutées ou ont une fréquence de décharge insuffisante pour que les fibres musculaires génèrent une force maximale . Au cours d’un exercice fatigant, une augmentation du pic de force surimposée peut être associée à une dégradation progressive de l’activation volontaire (pour une revue, voire ) et donc à la présence d’une fatigue d’origine centrale . Ainsi, Löscher et al. ont rapporté la présence d’une fatigue centrale lors d’une contraction isométrique des fléchisseurs plantaires (30 % MVC). Grâce à la stimulation électrique, Kent-Braun et Schillings et al. ont respectivement estimé à 20 et 12 % la part de la fatigue centrale dans la perte de force lors d’une contraction isométrique maximale des fléchisseurs dorsaux de la cheville et des fléchisseurs du bras. Par ailleurs, plusieurs travaux de l’équipe de Gandevia ont illustré, grâce à la stimulation magnétique transcrânienne (TMS), que la fatigue centrale pouvait contribuer pour plus de 25 % à la baisse de force produite lors de contractions maximales soutenues . Toutefois, la fatigue centrale semble contribuer de façon plus importante à la diminution de la production de force lors d’exercices de faible intensité . En effet, Smith et al. ont indiqué que les deux tiers de la fatigue pouvaient être attribués à des mécanismes centraux (supraspinaux) au cours de flexions isométriques de l’avant-bras à 5 % MVC.
Une des origines de la baisse de la commande centrale au cours d’exercices prolongés pourrait être la dégradation de l’excitation fournie par le cortex moteur . La mise en évidence de cette fatigue supraspinale est possible via la stimulation magnétique (ou électrique) transcrânienne . La TMS permet l’activation de neurones du cortex moteur qui induit la contraction des muscles qu’ils innervent . Pour étudier le développement de la fatigue supraspinale, on demande au sujet de réaliser, lors du maintien d’une contraction isométrique sous-maximale, de brèves contractions maximales à intervalles réguliers. Au cours de l’exercice, une augmentation de l’amplitude du pic de force induit par la TMS lors des contractions maximales indique alors la présence d’une fatigue d’origine supraspinale qui peut être attribuée à une excitation sous-maximale provenant du cortex moteur . Grâce à la TMS, plusieurs auteurs ont illustré une diminution de l’excitation fournie par le cortex moteur lors de flexions isométriques du bras soutenues à 15 et 30 % MVC . Par ailleurs, Soggard et al. ont estimé que la fatigue supraspinale représentait environ 40 % de la perte totale de force induite par une flexion isométrique du coude à 15 % MVC maintenue jusqu’à épuisement.
Les origines de la fatigue supraspinale sont mal identifiées. Cependant, plusieurs hypothèses explicatives ont été proposées. Tout d’abord, la fatigue supraspinale pourrait être liée à la déplétion ou l’accumulation de certains neurotransmetteurs cérébraux qui provoquerait une diminution de l’excitation descendante corticospinale. Le neurotransmetteur le plus étudié est la sérotonine (5-hydroxytryptamine [5-HT]). Newsholme et al. ont suggéré que l’exercice prolongé induisait une augmentation de l’activité sérotoninergique cérébrale qui limiterait la commande centrale et donc le recrutement des UM. Cela serait lié aux effets de la sérotonine sur la léthargie et la perte de motivation . La sérotonine ne peut pas franchir la barrière sanguine cérébrale. Les neurones cérébraux doivent donc la synthétiser eux-mêmes à partir de son précurseur le tryptophane (TRP). Au niveau plasmatique, le TRP libre entre en concurrence avec d’autres acides aminés (à chaîne branchée [AACB]) pour être transporté vers le cerveau. Ainsi, la synthèse de sérotonine cérébrale augmente avec une élévation du ratio plasmatique TRP libre/AACB. Cela se produirait à l’exercice prolongé car les AACB sont utilisés par les muscles pour produire de l’énergie (baisse des AACB) et parce que les acides gras libres, dont la concentration augmente, libèrent le TRP lié à l’albumine pour être transportés (augmentation du TRP libre). Plusieurs méthodes ont été utilisées pour mettre en évidence l’influence de l’activité sérotoninergique cérébrale dans la fatigue centrale et la performance. Les études portant sur l’ingestion de TRP et d’AACB ont fourni des résultats contradictoires sur l’influence de ces apports nutritionnels sur la durée de l’exercice . En revanche, l’apport d’hydrates de carbone limite l’activité du TRP et prolonge l’exercice lors d’une activité de pédalage à la puissance maximale aérobie . L’ingestion d’hydrates de carbone réduirait la lipolyse, la concentration d’acides gras libres circulant dans le plasma et l’augmentation induite par l’exercice de TRP libre, ce qui limiterait la synthèse de sérotonine cérébrale. Cela a été confirmé par de récents travaux qui ont démontré que l’ingestion d’hydrates de carbone au cours d’une épreuve de pédalage de trois heures à 200 W limitait la consommation de TRP cérébral et prévenait l’hypoglycémie . D’autres travaux sur la recapture de la sérotonine et sur l’administration de substances agonistes ou antagonistes de ce neurotransmetteur ont permis de confirmer que la sérotonine jouait un rôle important dans l’initiation du mouvement et la poursuite de l’exercice physique . La fatigue centrale pourrait également être influencée par l’activité d’autres neurotransmetteurs. La dopamine limiterait la synthèse de sérotonine en agissant sur l’enzyme clé de la synthèse de 5-HT, la TRP hydroxylase . Plus généralement, les catécholamines (adrénaline, noradrénaline, dopamine) pourraient avoir un effet sur la fatigue en raison de leur rôle sur la motivation et la motricité . D’ailleurs, l’amphétamine, une molécule analogue à la dopamine, améliore la performance physique . D’autres neurotransmetteurs comme le glutamate, l’acétylcholine, l’adénosine et l’acide gamma-amino butyrique (GABA) ont été proposés comme agissant dans le développement de la fatigue centrale . Les interactions se produisant entre ces neurotransmetteurs pourraient également influencer la fatigue centrale . Cependant, trop peu de données publiées sont disponibles pour tirer des conclusions claires sur l’influence de ces neurotransmetteurs dans l’avènement de la fatigue. Aussi, les variations de la concentration des différents neurotransmetteurs pourraient également altérer la réponse cognitive ou être impliquées dans les mécanismes subconscients de la fatigue .
Ensuite, la commande centrale pourrait être limitée par les variations de concentration cérébrale d’autres substances comme l’ammonium ou le glycogène. Au cours de l’exercice, la concentration plasmatique en ammonium augmente, principalement à cause de la désamination des acides AACB . L’ammonium peut facilement franchir la barrière cérébrale sanguine et son accumulation pourrait alors être un facteur de la baisse de l’activité du cortex moteur lors d’exercices prolongés de par ses effets sur le flux sanguin cérébral, l’activité de certains neurotransmetteurs et sur la transmission synaptique . Le glycogène cérébral pourrait aussi jouer un rôle dans la fatigue centrale car l’activation cérébrale est associée à une baisse des réserves en glycogène cérébral . Or, les réserves de glycogène cérébral peuvent être rapidement épuisées en raison de leur petite quantité. Bien que ces réserves soient fréquemment renouvelées, leur déplétion pourrait influencer la fonction cérébrale et, notamment, avoir une incidence sur l’activité de la sérotonine . L’influence de l’activité des neurotransmetteurs et des concentrations cérébrales en ammonium et en glycogène sur la fatigue centrale et la performance a été mise en évidence pour des exercices prolongés de longue durée (> 30 minutes) . De la même façon, l’hyperthermie, phénomène parfois associé à l’exercice sportif, réduirait l’activité de la commande nerveuse centrale . Enfin, il émerge une nouvelle hypothèse relative à l’implication des signaux humoraux (cytokines) dérivés de l’activité musculaire en présence de fatigue . Néanmoins, les mécanismes liant les cytokines à la fatigue demeurent encore mal compris et nécessitent de nouvelles recherches.
Une autre hypothèse concernant la fatigue supraspinale suggère que certaines afférences musculaires, relatives à l’état biochimique et à la capacité de production de force des muscles, limitent l’activité corticale. Grâce à une fatigue induite par électrostimulation de l’interosseous, Pitcher et Miles ont démontré que la fatigue supraspinale, illustrée par la diminution des potentiels moteurs évoqués (MEP), pouvait être modifiée par les afférences musculaires. Toutefois, ces auteurs ne statuent pas sur le fait que cette fatigue supraspinale survienne au niveau du cortex moteur ou en amont de celui-ci. Grâce à l’analyse de nombreux travaux, Gandevia rassemble la preuve d’une inhibition supraspinale. Cet auteur indique que les feedbacks musculaires renseignant sur l’état biochimique et les capacités de production de force sont susceptibles de dégrader la stimulation provenant des sites corticaux. Le rôle des afférences musculaires et l’association entre fatigue périphérique et fatigue supraspinale peuvent être démontrés en empêchant la récupération du muscle fatigué à la fin de la contraction fatigante par ischémie . À l’arrêt de la contraction volontaire, les modifications induites par la fatigue au niveau de la commande nerveuse peuvent être « récupérées », alors que l’ischémie maintient les décharges des afférences musculaires III et IV sensibles aux métabolites produites lors de la fatigue. La mesure de l’activation volontaire par TMS montre alors que la fatigue supraspinale ne se restaure pas tant que le muscle reste sous ischémie . Au contraire, les activités corticospinale et motoneuronale semblent revenir à des valeurs normales . Cela suggère qu’un mécanisme, en relation avec l’état de fatigue du muscle, agit de façon à limiter son activation volontaire, mais cela ne semble pas se produire au niveau des motoneurones ou de l’excitation motrice corticale. Il est alors possible que les afférences musculaires sensibles à la fatigue agissent « en amont » du cortex moteur pour limiter la commande volontaire , ce qui précise les résultats de Pitcher et Miles .
Au niveau spinal, la fatigue centrale pourrait être associée à plusieurs mécanismes. Tout d’abord, les afférences inhibitrices provenant de récepteurs intramusculaires semblent impliquées dans la baisse de l’activité du motoneurone. En effet, Bigland-Ritchie et al. ont montré que la fréquence de décharge des motoneurones pouvait être régulée par des réflexes périphériques en réponse aux variations métaboliques musculaires induites par la fatigue. Ainsi, les afférences musculaires de type III et IV (métaborécepteurs) semblent être stimulées en condition d’ischémie , d’hypoxémie et d’accumulation extracellulaire de potassium (K + ) et de lactate . La stimulation de ces métaborécepteurs au cours d’un exercice fatigant inhiberait alors l’activité des motoneurones alpha . Les fuseaux neuromusculaires (afférences Ia et II), disposés parallèlement aux fibres musculaires, renseignent le système nerveux sur la longueur et les variations de longueur du muscle . Macefield et al. ont rapporté que la fréquence de décharge de ces récepteurs diminuait progressivement au cours d’une contraction isométrique inférieure à 30 % MVC. Cette baisse était importante au début de l’exercice, puis la diminution, se prolongeait plus lentement. La chute de la fréquence de décharge induite par les fuseaux neuromusculaires serait alors associée à une limitation de l’activité motoneuronale alpha . Par ailleurs, la sensibilité de ces fuseaux neuromusculaires peut être réduite par des modifications de la structure du muscle (e.g. variations de la raideur musculotendineuse) pouvant être induites par des contractions ou des étirements répétés . Les modifications de la raideur musculaire pourraient également influencer l’activité électrique musculaire et contribuer à la fatigue . Néanmoins, il reste à déterminer de quelle façon ces changements de raideur pourraient influencer l’activité des muscles en contraction.
Les organes tendineux de Golgi (afférences Ib), situés au niveau des jonctions myotendineuses et musculoaponévrotiques, renseignent le SNC sur les tensions produites au sein du muscle . Ces mécanorécepteurs sont supposés inhiber l’activité neuronale . L’action des organes tendineux de Golgi sur l’activité des motoneurones lors d’une tâche fatigante est délicate à déterminer car ces afférences sont difficiles à isoler et leurs projections incluent des interneurones recevant des signaux provenant des afférences Ia . Toutefois, l’influence des signaux émis par les fuseaux neuromusculaires et les organes tendineux de Golgi reste peu étudiée et est incertaine, principalement car leurs effets peuvent être atténués au niveau spinal et qu’ils sont de moindre importance par rapport à ceux des nombreuses afférences III et IV .
Ensuite, la baisse de l’activité du motoneurone pourrait aussi être due à des capacités intrinsèques de celui-ci . D’après ces auteurs, le motoneurone pourrait adapter sa fréquence de décharge au cours de la fatigue en réponse à une excitation constante. La stimulation des afférences III et IV pourrait induire des changements des propriétés intrinsèques de la membrane du motoneurone qui modifierait sa fréquence de décharge . Cette baisse de la fréquence de décharge ( late adaptation ) serait alors plus prononcée pour les fibres rapides comparées aux fibres lentes . Cependant, les connaissances sur ce phénomène restent limitées et ont été observées chez l’animal.
Par ailleurs, l’inhibition intracorticale pourrait également être un mécanisme impliqué dans la baisse de performance musculaire en condition fatigante. Récemment, McNeil et al. ont suggéré que, lors d’une contraction maximale des fléchisseurs du coude maintenue durant deux minutes, l’inhibition intracorticale augmentait de façon concomitante à la progression de la fatigue.
Enfin, les motoneurones (principalement ceux des UM rapides) reçoivent une inhibition provenant des cellules de Renshaw, qui sont stimulées par ces mêmes motoneurones et par des influences descendantes et périphériques. À l’aide de la méthode indirecte du réflexe de Hoffmann (réponse musculaire induite par l’excitation des afférences Ia lors d’une stimulation électrique), plusieurs études ont suggéré que cette inhibition augmentait au cours d’efforts maximaux , mais diminuait lors de contractions sous-maximales à 20 % MVC associées à la présence d’une fatigue centrale .
Les sites et mécanismes physiologiques affectés par la fatigue neuromusculaire au niveau central sont donc nombreux. Celle-ci semble perturber également le fonctionnement de processus physiologiques dans la partie « périphérique » du système neuromusculaire.
2.2.2
Facteurs périphériques de la fatigue
Les facteurs d’origine périphérique de la fatigue regroupent les altérations pouvant survenir lors de la transmission neuromusculaire, de la propagation des potentiels d’actions musculaires, du couplage excitation-contraction et des mécanismes contractiles associés ( Fig. 1 ).
La transmission neuromusculaire désigne le transfert du potentiel d’action (PA) nerveux en potentiel d’action musculaire (PAM) et se déroule au niveau de la JNM. Au cours de la fatigue, ce mécanisme peut être altéré par :
- •
une propagation du potentiel nerveux insuffisante au niveau des terminaisons axonales ;
- •
une défaillance du couplage excitation-sécrétion des neurotransmetteurs au niveau de la fente synaptique ;
- •
une déplétion des neurotransmetteurs ;
- •
une réduction des neurotransmetteurs libérés ;
- •
une diminution de la sensibilité des récepteurs post-synaptiques à acétylcholine et de la membrane .
Lors d’une contraction maintenue, la fatigue induirait une baisse de l’excitabilité des axones de petit diamètre. Une légère diminution de l’excitation aboutirait alors à une inactivation de ces axones et à une baisse de la quantité de neurotransmetteurs libérés dans la fente synaptique . Ces auteurs ont rapporté, in vitro, que certaines terminaisons axonales des motoneurones innervant le diaphragme du rat n’étaient plus activées après une stimulation électrique musculaire de plusieurs minutes. Ce phénomène n’était pas dépendant de la concentration du bain physiologique en K + , calcium (Ca 2+ ) ou hydrogène (H + ), les auteurs ont alors conclu à l’influence de la concentration en oxygène musculaire sur la propagation du PA. De plus, l’amplitude des potentiels de plaque motrice diminue au cours d’un exercice fatigant en raison de la baisse de la quantité de neurotransmetteurs (acétylcholine) libérés par chaque terminaison axonale. Cela serait en relation avec une réduction du nombre de vésicules d’exocytose et/ou une baisse de la quantité d’acétylcholine libérée par vésicule . Par ailleurs, les travaux de Van Lunteren et Moyer ont suggéré que les variations de concentration intracellulaire en Ca 2+ au niveau des terminaisons nerveuses jouaient un rôle clé (via l’exocytose) dans la quantité de neurotransmetteurs libérés. Toutefois, ces travaux ont été réalisés in vitro chez l’animal, ce qui limite les conclusions sur l’implication des phénomènes présentés lors de mouvements volontaires chez l’homme. L’efficacité de la transmission neuromusculaire est également dépendante de la sensibilité à l’ACh des récepteurs membranaires post-synaptiques. Or, une exposition prolongée des récepteurs cholinergiques à l’ACh semble les désensibiliser à ce neurotransmetteur et se traduire par un allongement du temps nécessaire à la liaison de l’ACh sur les récepteurs . Cependant, l’importance de la désensibilisation des récepteurs post-synaptiques au sein du processus de fatigue, tout comme les autres mécanismes pouvant induire une dégradation de la transmission neuromusculaire, n’a pas été établie de façon certaine. La dégradation de la transmission neuromusculaire peut être illustrée en provoquant par stimulation électrique des ondes M avant, pendant et après une contraction fatigante. L’onde M correspond à l’activité électrique musculaire mesurée par électromyographie en réponse à une stimulation électrique du nerf . Une baisse de l’amplitude de l’onde M peut alors être interprétée comme une dégradation d’un ou plusieurs mécanismes impliqués dans le passage du PA axonal vers le PAM. La propagation du signal sur le sarcolemme influence alors de façon prépondérante l’amplitude de l’onde M. Toutefois, les résultats obtenus grâce à cette technique de stimulation électrique sont discutés dans la littérature et leur pertinence, dans certaines circonstances, remise en question . En effet, les caractéristiques de l’onde M peuvent être affectées par certains facteurs tels que la synchronisation des potentiels d’action des fibres musculaires et/ou le degré de dispersion des neurotransmetteurs lors de leur libération . Par ailleurs, les changements de l’onde M ne reflèteraient pas les variations des potentiels d’action intracellulaires . À ce sujet, certains auteurs ont mis en avant les avantages de l’EMG à haute densité qui, à l’aide de matrices d’électrodes, permet d’ajouter une information spatiale à l’information temporelle contenue dans le signal EMG .
Les résultats relatifs aux effets de la fatigue sur les changements de la propagation du signal sont variables . Ainsi, certains travaux rapportent que la dégradation de la transmission neuromusculaire apparaît comme un facteur limitant l’excitation du muscle et contribuerait à la baisse de production de force qui semble survenir de façon plus importante pour des exercices de faible intensité et de longue durée que lors de contractions intenses et brèves . Néanmoins, d’autres travaux n’ont observé aucun changement de l’onde M suite à des contractions sous-maximales de durées inférieures à cinq minutes . Il apparaît donc difficile de tirer des conclusions quant à l’influence de la transmission neuromusculaire sur la production de force en condition fatigante.
Les potentiels d’action musculaires sont générés lorsque la somme des potentiels présynaptiques (excitateurs et inhibiteurs) atteint ou dépasse le seuil d’excitabilité de la cellule musculaire. Toutefois, plusieurs facteurs sont susceptibles d’altérer la transformation de l’excitation nerveuse en force musculaire et les exercices fatigants peuvent y contribuer . Il s’agit de la propagation du PAM le long du sarcolemme et des tubules transverses, de la perméabilité du réticulum sarcoplasmique (RS) aux ions Ca 2+ , du mouvement des ions Ca 2+ à l’intérieur du sarcoplasme, du repompage du Ca 2+ par le RS, de la liaison du Ca 2+ à la troponine et de l’interaction entre la myosine et l’actine associée au travail des ponts d’union. Le Ca 2+ joue un rôle essentiel dans les mécanismes à l’origine de la production de force. Westerblad et Allen ont observé que lors d’une contraction isométrique prolongée, la force développée diminuait, dans un premier temps, alors que la concentration intramusculaire en Ca 2+ était élevée, suggérant une dégradation de la production de force au niveau des myofibrilles. Dans un second temps, la baisse de force était associée à une baisse de la quantité de Ca 2+ libéré par le RS.
Parmi les nombreux changements métaboliques associés à la contraction musculaire prolongée, deux principaux facteurs semblent être à l’origine de la diminution de la capacité de production de force des myofibrilles . Il s’agit des augmentations de la concentration intracellulaire en ions hydrogène (H + ) et en phosphate inorganique (Pi). La concentration en Pi s’élève au cours de l’exercice en raison de la dissociation de la phosphocréatine en Pi + créatine et de l’hydrolyse de l’ATP, alors que les ions H + s’accumulent en relation avec l’hydrolyse de l’ATP et la production d’acide lactique (qui se produit quand l’activité glycolytique dépasse la capacité oxydative des mitochondries ou quand l’apport d’oxygène est limité). L’accumulation de H + a longtemps été considérée comme une cause importante de la baisse de production de force des myofibrilles car elle est associée à une diminution du pH intracellulaire qui perturbe le déroulement des réactions chimiques. Néanmoins, ces résultats ont été obtenus pour des températures inférieures aux températures physiologiques. En effet, certains auteurs ont montré que l’acidification du milieu avait un effet très limité sur les capacités de production de force à des températures physiologiques . Si l’acidose n’est pas la cause de la chute de production de force, celle-ci devrait être due à l’accumulation de Pi. Il a été établi, à basse température (10–15°), qu’une augmentation intracellulaire de Pi limitait la force produite quand le milieu était saturé en Ca 2+ . Cependant, les travaux ayant testé les variations de température présentent des résultats contradictoires quant à l’influence des Pi sur la force produite . Depuis, l’influence du Pi sur les mécanismes de production de force a été prouvée en réduisant sa concentration dans le milieu, ce qui induisait une augmentation de la force . Le Pi pourrait perturber la production de force en diminuant la sensibilité des myofibrilles au Ca 2+ et en agissant directement au niveau des ponts d’union . En effet, l’augmentation de la concentration en Pi en condition de fatigue perturberait le déroulement des cycles de contraction-relâchement des ponts d’union . L’ensemble de ces résultats suggèrent que la diminution de la force produite, observée dans les premiers instants de la contraction alors que la concentration intracellulaire en Ca 2+ est élevée, serait due à l’accumulation de Pi.
Ensuite, lorsque l’exercice musculaire se poursuit, la baisse de production de force serait liée à une diminution de la quantité de Ca 2+ libéré par le RS. Ce phénomène se produirait plus tardivement au sein des fibres ayant de fortes capacités oxydatives et plus rapidement pour des fibres fatiguées en conditions anaérobies . Les variations de concentration en Pi et en adénosine triphosphate (ATP) sembleraient à l’origine de la dégradation de la libération du Ca 2+ . L’accumulation du Pi au sein de la cellule musculaire induite par l’exercice favoriserait sa pénétration dans le RS. Le Pi se lierait alors aux ions Ca 2+ pour donner du phosphate de calcium (Ca 2+ -Pi), limitant ainsi la quantité de Ca 2+ présente dans les citernes du RS et pouvant être libérée dans le sarcoplasme . Par ailleurs, la quantité de Ca 2+ libéré par le RS semble associée à l’activité de l’ATP. En effet, quatre sites participant à la libération du Ca 2+ sont sensibles aux concentrations d’ATP . Tout d’abord, la libération du Ca 2+ est dépendante du cheminement du PAM du sarcolemme vers les tubules transverses jusqu’au RS. Au cours d’une contraction prolongée, la succession de potentiels d’action provoque un déséquilibre des concentrations ioniques en Na + et K + de part et d’autre de la membrane cellulaire. Les pompes Na + /K + ATPasiques utilisent l’énergie fournie par l’hydrolyse de l’ATP afin de transporter trois ions Na + à l’extérieur de la cellule et faire entrer deux ions K + pour restaurer l’homéostasie cellulaire. Une altération de l’activité de ces pompes (e.g. baisse de concentration en ATP) induirait alors une perturbation de l’excitabilité de la membrane qui altérerait la propagation du PA vers le RS et par conséquent, la libération du Ca 2+ . Ensuite, l’ouverture des canaux à K + du sarcolemme est liée à la concentration cellulaire en ATP . L’ouverture de ces canaux, induite par la baisse de la concentration en ATP associée à l’exercice musculaire, provoque une sortie d’ions K + vers l’extérieure de la cellule musculaire. De la même façon que présentée précédemment, ce mouvement ionique pourrait altérer les PA et limiter la libération de Ca 2+ . Toutefois, ces canaux ne semblent pas agir de façon significative au cours de la fatigue . Lors de la contraction musculaire, le Ca 2+ est libéré par le RS pour aller se fixer à la troponine et permettre la contraction musculaire. Ensuite, le Ca 2+ est repompé au sein de cette structure afin d’être à nouveau disponible. Ainsi, une dégradation de l’activité des pompes ATPasiques à Ca 2+ participant au réacheminent du Ca 2+ vers le RS, induirait une baisse de la quantité de Ca 2+ présent dans cette structure et moins de Ca 2+ pourrait être libéré. Cependant, l’activité de ces pompes ne semble pas être à l’origine de la dégradation de la libération du Ca 2+ . Enfin, la transmission du signal entre les tubules transverses et le RS et la libération du Ca 2+ sont également dépendantes des réserves en ATP. La fatigue, et la baisse des réserves en ATP, induirait alors une moindre phosphorylation de certains sites impliqués dans le cheminement du PA et pourrait limiter la libération de Ca 2+ , notamment au niveau des canaux à Ca 2+ du RS .
Les ions magnésium (Mg 2+ ) pourraient également être impliqués dans la dégradation de la libération du Ca 2+ car ils interviennent dans le fonctionnement du RS. En effet, lors de l’activation musculaire, une augmentation de la concentration en Mg 2+ dans le sarcoplasme réduit la libération du Ca 2+ par le RS . Par ailleurs, Westerblad et Allen ont montré que, lors de l’exercice, les concentrations intracellulaires de Mg 2+ augmentaient, ce qui pourrait induire une baisse de la force du muscle. Cette augmentation de Mg 2+ au cours de l’exercice serait en partie due au fait que les ions Mg 2+ sont liées aux molécules d’ATP (dégradées lors de l’exercice). L’activation des canaux calciques du RS libèrent également des ions Mg 2+ et pourrait participer à l’augmentation de leur concentration .
Enfin, la diminution de la quantité de Ca 2+ libérée par le RS pourrait également avoir d’autres origines . Ainsi, il a été reporté que des mitochondries du muscle squelettique contenaient plus de Ca 2+ après un exercice exhaustif que des mitochondries d’un muscle contrôle . Ainsi, la capture de Ca 2+ par les mitochondries pourrait participer à la baisse de Ca 2+ disponible. Certains travaux ont aussi évoqué la possibilité que la fatigue musculaire provoquée par un exercice excentrique induirait une rupture mécanique entre les tubules-t et le RS menant à une dégradation du couplage excitation-contraction . Ce dérèglement susciterait une libération inadéquate de Ca 2+ du RS. Un tel découplage excitation-contraction pourrait expliquer la fatigue à basse fréquence de longue durée que l’on observe à la suite de diverses formes d’exercice physique .
L’ensemble de ces travaux indiquent que la baisse de la production de force lors d’un exercice soutenu est principalement liée à une baisse de la quantité de Ca 2+ libéré par le RS. Cela serait principalement dû à l’accumulation intracellulaire de Pi et à la diminution des réserves en ATP associées à l’exercice fatigant.
Lors d’un exercice soutenu, l’apport sanguin et les réserves en substrats métaboliques sont également susceptibles d’influencer la performance . Au cours d’une contraction musculaire, la dégradation du flux sanguin a été l’un des premiers mécanismes identifié comme participant à la fatigue. Lors de l’activité physique, une augmentation du flux sanguin est nécessaire pour apporter des substrats aux muscles actifs, évacuer les métabolites et dissiper la chaleur. Cependant, la contraction musculaire engendre une compression des vaisseaux sanguins et, par conséquent, une diminution de l’apport sanguin aux muscles actifs pouvant aller jusqu’à provoquer une ischémie complète en régime de contraction isométrique. Si la contraction se prolonge, l’apport sanguin limité aura pour incidence un apport moindre en oxygène et favorisera l’activité des voies métaboliques anaérobies. La baisse du flux sanguin va provoquer une accumulation plus rapide des métabolites associés à la contraction musculaire (e.g. Pi, H + ), et donc accélérer le processus de fatigue et la baisse de production de force. Sjogaard et al. ont montré que l’apport sanguin diminuait avec l’augmentation de l’intensité de l’exercice lors d’extensions de la jambe à des intensités comprises entre 5 et 50 % MVC. En outre, plus l’intensité de la contraction est importante, plus l’ischémie sera précoce . À ce sujet, Crenshaw et al. ont montré que le temps de maintien d’un exercice impliquant les extenseurs de la jambe à 25 % MVC coïncidait avec la durée nécessaire pour provoquer une ischémie complète . Néanmoins, l’apport sanguin reste vraisemblablement inchangé pour des contractions menées à moins de 15 % MVC .
Dans la majorité des contractions musculaires, la disponibilité en substrats métaboliques ne constitue pas un facteur limitant la performance. Cependant, lors de certaines activités de longue durée comme le pédalage à intensité sous-maximale (70 à 80 % de la puissance maximale aérobie), l’incapacité à maintenir la force nécessaire à la réalisation de la tâche coïnciderait avec la déplétion des réserves de glycogène des extenseurs de la jambe . D’ailleurs, un apport supplémentaire en glucose permet aux sujets de maintenir l’exercice plus longtemps .
Toutefois, certains auteurs suggèrent que ce ne serait pas la fatigue centrale et/ou périphérique induite par l’exercice qui provoquerait l’arrêt d’exercices aérobies d’intensité élevée, mais plutôt le désengagement du sujet de la tâche en lien avec la perception de l’effort . La validité de ce modèle psychobiologique reste, néanmoins, à valider.
2.3
Adaptations à la fatigue
La fatigue neuromusculaire (centrale et périphérique) va provoquer des adaptations du système neuromusculaire visant au maintien de la production de force ( Fig. 2 ). Ce chapitre traite des principaux mécanismes adaptatifs impliqués lorsque la fatigue survient au niveau du système neuromusculaire au cours d’exercices maximaux et sous-maximaux de durée courte à moyenne. Les mécanismes décrits sont de trois types :
- •
la modulation de l’activité des UM ;
- •
l’augmentation de l’efficacité de l’activité musculaire (i.e., potentialisation) ;
- •
le mécanisme « protecteur » dit de sagesse musculaire.
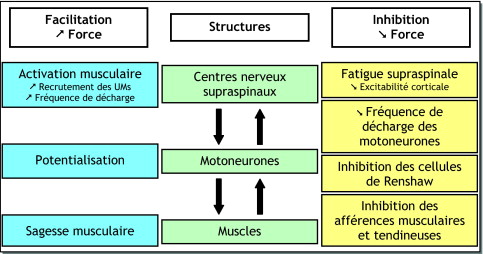
Related posts:
 Self care programs and multiple sclerosis: Physical therapeutics treatment – literature review
Translation into Arabic and validation of the ASES index in assessment of shoulder disabilities
Risk assessment analysis of the future technical unit dedicated to the evaluation and treatment of motor disabilities
Neuromuscular diseases
Traumatic brain injury
Allografts: Graft Sterilization and Tissue Banking Safety Issues
Self care programs and multiple sclerosis: Physical therapeutics treatment – literature review
Translation into Arabic and validation of the ASES index in assessment of shoulder disabilities
Risk assessment analysis of the future technical unit dedicated to the evaluation and treatment of motor disabilities
Neuromuscular diseases
Traumatic brain injury
Allografts: Graft Sterilization and Tissue Banking Safety Issues
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


