Neuroendocrine Disorders
Thomas Moshang Jr.
Adda Grimberg
Neuroendocrinology, the study of the body’s homeostatic control mechanisms, examines the interactions between the nervous and endocrine systems. Both neurons and endocrine cells exert their actions through release of chemical messengers that bind to specific cell receptors, although the former classically release messengers across synapses and the latter into the circulation. Both systems seem to converge in the hypothalamic–pituitary unit, sometimes referred to as the body’s master gland. Neuroendocrine disorders in children disturb growth, sexual maturation and function, water balance, and adrenal and thyroid functions. The rising number of childhood cancer survivors constitutes an increasingly important subgroup of children with neuroendocrine disorders due to the sequelae of radiotherapy and chemotherapy.
NORMAL CONTROL MECHANISMS: ANATOMY AND PHYSIOLOGY
The hypothalamic–pituitary unit is located at the base of the third ventricle where the hypothalamus receives multiple afferent neural connections from higher brain centers. Also, chemical agents from the cerebrospinal fluid are transmitted from the third ventricle to hypothalamic axon terminals and capillaries via tanycytes, bipolar ependymal cells. The hypothalamus communicates with the anterior pituitary gland through a special portal circulation that not only is fenestrated (the exception to the blood–brain barrier) but that also transmits information bidirectionally. The link to the posterior pituitary is a direct physical connection; the axons of the posterior pituitary actually have their cell bodies in the hypothalamic nuclei (Fig. 377.1).
Afferent signals from the brain and hypothalamus stimulate pituitary hormone secretion, which in turn activates hormone synthesis and release by other endocrine glands in the body. These hormones provide quantitative data to the hypothalamic–pituitary unit to determine the need for ongoing activity (positive feedback) or termination of signal (negative feedback). An extra step of modulation occurs in the anterior pituitary, as the hypothalamus secretes releasing and inhibitory factors (e.g., growth hormone–releasing hormone [GHRH] and somatostatin) into the portal network to control activity of the anterior pituitary hormones: growth hormone (GH), thyroid-stimulating hormone (TSH), the gonadotropins (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]), adrenocorticotropin hormone (ACTH), and prolactin. Vasopressin (antidiuretic hormone [ADH]) and oxytocin, the two hormones secreted by the posterior pituitary, are synthesized by cell bodies within the hypothalamic nuclei and transferred in secretory granules via axonal transport down the pituitary stalk (Table 377.1; see Fig. 377.1).
NEUROENDOCRINE DISORDERS
Neuroendocrine disorders may present at any age with variable clinical manifestations dependent on the cause and the hormones that are produced or deficient (Table 377.2). Growth failure is the most common presentation of neuroendocrine disturbances in children. It may arise from deficiencies of several pituitary hormones, either in combination or singly, or from loss of growth signals from the hypothalamus and higher brain centers. Thus, accurate and consistent plotting of a child’s growth curve is often the pediatrician’s first clue to an underlying neuroendocrine problem.
Congenital or Genetic Forms of Neuroendocrine Disease
Congenital or genetic forms of neuroendocrine disorders may present in the newborn period or later in life. The genetic causes of a number of these disorders are known, including mutations in the GHRH receptor, GH gene, Pit1 gene, Prop1 gene, Kallmann syndrome (hypogonadotropism with anosmia), and others. It is beyond the scope of this section to discuss the details of these gene mutations, but the clinical aspects are presented here. Parks et al. recently reviewed the heritable disorders of pituitary development.
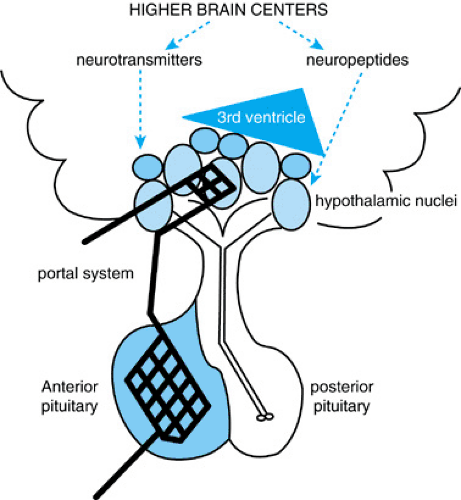 FIGURE 377.1. Anatomy of communication within the hypothalamic–pituitary unit. Input to the hypothalamic nuclei originates from higher brain centers, the cerebrospinal fluid in the third ventricle, and the anterior pituitary through a special bidirectional fenestrated portal circulatory network. The hypothalamic nuclei affect anterior pituitary functioning through releasing and inhibitory factors transmitted by the portal capillaries and posterior pituitary functioning by axonal transport. |
TABLE 377.1. OVERVIEW OF THE NEUROENDOCRINE AXES AND THEIR FEEDBACK LOOPS* | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Disorders of Growth
Congenital neuroendocrine growth disorders are caused by deficiency of hypothalamic–pituitary hormone(s) or abnormality of hormone receptor(s), and all are probably caused by gene mutations, although some of the genetic defects have not yet been elucidated.
TABLE 377.2. NEUROENDOCRINE DISORDERS IN CHILDREN* | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Panhypopituitarism
Congenital panhypopituitarism, or the deficiency of more than one pituitary hormone, is caused by gross malformation and histologic or molecular abnormalities of the hypothalamic–pituitary unit. Gross hypothalamic–pituitary malformation may be associated with other midline brain malformations, including holoprosencephaly and anencephaly. Holoprosencephaly, characterized by ventral midline fusion of forebrain structures, is the most frequent brain malformation in humans. Because induction of hypothalamic development is intimately involved in forebrain midline signaling
during embryogenesis, mild cases of holoprosencephaly may present clinically with only hypothalamic/pituitary dysfunction. Four genes have been shown to cause holoprosencephaly in humans when mutated, including sonic hedgehog (SHH). Septooptic dysplasia, or de Morsier syndrome, describes the association of optic nerve hypoplasia with septum pellucidum agenesis; additional associations may be schizencephaly or callosal absence, and a case of septooptic dysplasia with cerebrocortical dysplasia has been reported. Septooptic dysplasia occurs with variable degrees of neuroendocrine dysfunction, primarily hypothalamic involvement and GH deficiency. Septooptic dysplasia has been associated with mutations of the HESX1 homeobox gene. Thus, mutations in transcription factors active during early hypothalamic–pituitary development frequently result in gross abnormalities that may be visualized with brain imaging. Mutations in factors active later during cellular differentiation and function (such as Pit1 or PROP1) may result in histologic and functional changes, but not radiologically apparent abnormalities. Nonetheless, the clinical characteristics are similar.
during embryogenesis, mild cases of holoprosencephaly may present clinically with only hypothalamic/pituitary dysfunction. Four genes have been shown to cause holoprosencephaly in humans when mutated, including sonic hedgehog (SHH). Septooptic dysplasia, or de Morsier syndrome, describes the association of optic nerve hypoplasia with septum pellucidum agenesis; additional associations may be schizencephaly or callosal absence, and a case of septooptic dysplasia with cerebrocortical dysplasia has been reported. Septooptic dysplasia occurs with variable degrees of neuroendocrine dysfunction, primarily hypothalamic involvement and GH deficiency. Septooptic dysplasia has been associated with mutations of the HESX1 homeobox gene. Thus, mutations in transcription factors active during early hypothalamic–pituitary development frequently result in gross abnormalities that may be visualized with brain imaging. Mutations in factors active later during cellular differentiation and function (such as Pit1 or PROP1) may result in histologic and functional changes, but not radiologically apparent abnormalities. Nonetheless, the clinical characteristics are similar.
Common clinical findings in the newborn with panhypopituitarism include hypoglycemia and jaundice and, in boys, micropenis. Newborn screening for hypothyroidism may detect a low thyroxine level (T4) with a normal TSH level. In infants with gross malformations, there may be midline defects (such as cleft palate), neurologic signs, and visual defects (especially nystagmus). In the older child, there may be a single central incisor or, most commonly, growth failure. Treatment consists of replacing the identified deficient hormones and addressing the underlying cause.
Isolated Growth Hormone Deficiency
Isolated GH deficiency (GHD) occurs as a spectrum of clinical diseases, all sharing growth failure as the main phenotypic trait. The familial forms include autosomal recessive and autosomal dominant patterns of inheritance. Complete or classic GHD is not difficult to diagnose. However, partial GHD or insufficiency is often more difficult to confirm biochemically because of the hormone’s circadian rhythm. Indirect means of measuring GH function have therefore been devised, including random growth factor (insulinlike growth factor-1 [IGF-1] and IGF-binding protein [IGFBP]-3) levels, provocative GH testing, and overnight serum GH sampling. Because all of these tests have questionable specificity, interassay differences, or, in the case of GH, paucity of published age-, gender-, or pubertal-specific normal ranges, diagnosis of GHD cannot rely on any single parameter. The combination of subnormal growth velocity, delayed bone age, and two provocative stimulation tests with failing results is accepted as adequate indication of GHD. Provocative GH testing falsely passes children with neurosecretory GHD; the pharmacologic stimulus bypasses the defect and prompts release of normal levels of GH by the unaffected pituitary gland, whereas spontaneous GH secretion in these children is blunted because of the defect in the hypothalamic or higher brain centers’ triggering of GH release. Clinical concern based upon the knowledge that neurosecretory GHD exists, especially after cranial irradiation, can be diagnosed by overnight GH sampling. The various neuromodulators of GH secretion are listed in Table 377.3. Children with GH insufficiency, including neurosecretory GHD, achieve improvements in final height with GH therapy, as do children with classic GHD but not children with non-GHD short stature.
Disorders of Sexual Maturation and Function
Isolated Gonadotropin Deficiency
Isolated gonadotropin deficiency actually may be more common than GHD. However, most patients present to reproductive endocrinologists with
the chief complaint of infertility. More complete deficiency of gonadotropins presents as inadequate adolescent sexual development. Distinguishing idiopathic hypogonadotropic hypogonadism from constitutional delay of growth and development as well as from psychological dysfunction (depression or anorexia nervosa) can be difficult. Gonadotropin-releasing hormone (GnRH) testing, evaluating the gonadotropin response to an administered GnRH bolus, may be misleading, because the latter conditions also do not demonstrate a gonadotropin response to GnRH. Partial gonadotropin deficiency, conversely, may proceed slowly through puberty and a GnRH bolus testing will cause release of gonadotropins. Patients with Kallmann syndrome (clinically manifested by delayed puberty associated with anosmia) are often easier to diagnose because of the anosmia. This is a familial syndrome, and genetic analysis often detects a mutation in the X chromosome KAL locus that encodes a developmental chemoattractant for both GnRH-secreting and olfactory neuron migration.
the chief complaint of infertility. More complete deficiency of gonadotropins presents as inadequate adolescent sexual development. Distinguishing idiopathic hypogonadotropic hypogonadism from constitutional delay of growth and development as well as from psychological dysfunction (depression or anorexia nervosa) can be difficult. Gonadotropin-releasing hormone (GnRH) testing, evaluating the gonadotropin response to an administered GnRH bolus, may be misleading, because the latter conditions also do not demonstrate a gonadotropin response to GnRH. Partial gonadotropin deficiency, conversely, may proceed slowly through puberty and a GnRH bolus testing will cause release of gonadotropins. Patients with Kallmann syndrome (clinically manifested by delayed puberty associated with anosmia) are often easier to diagnose because of the anosmia. This is a familial syndrome, and genetic analysis often detects a mutation in the X chromosome KAL locus that encodes a developmental chemoattractant for both GnRH-secreting and olfactory neuron migration.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


