Nephrogenic Diabetes Insipidus
Arundhati S. Kale
L. Leighton Hill
Nephrogenic diabetes insipidus (NDI) is a hereditary or acquired disorder characterized by renal tubular resistance to the antidiuretic hormone arginine vasopressin (AVP). The inability to concentrate the urine because of this tubular disease leads to marked polyuria with compensatory polydipsia.
Inheritance of the hereditary forms of NDI may be X-linked or autosomal. Approximately 90% of congenital NDI cases are caused by mutations of the AVP receptor 2 gene (AVPR2), which codes for the vasopressin V2 receptor and occurs predominantly in males, consistent with an X-linked inheritance, with the locus at Xq28. Autosomal-recessive and, rarely, autosomal-dominant NDI is caused by mutations of the aquaporin 2 gene (AQP2), which has been localized to chromosome region 12q13. In the X-linked cases, at least 155 mutations have been identified that lead to the defective intracellular transport of receptors, thus resulting in the nonexpression of the receptors for binding AVP at the cell surface. Females usually show a limited tubular response to vasopressin, and even in males, the degree of severity of the defect varies; most males are diagnosed in early infancy because of a severe defect, but some escape detection until the second or third decade of life. Hereditary NDI in males with the severe defect usually is more severe than the acquired type, which has two general causes: loss of the concentration gradient in the medullary interstitial tissues or decreased responsiveness of the distal tubules and collecting ducts to AVP. The former occurs as a result of the tissue destruction occurring with obstructive uropathy, vesicoureteral reflux, sickle cell nephropathy, cystic disease, pyelonephritis, interstitial nephritis, or nephrocalcinosis. Decreased responsiveness of the distal tubules and collecting ducts to AVP can be caused by hypokalemia, hypercalcemia, amyloidosis, sarcoidosis, and various drugs that interfere with the action of AVP, such as lithium, demeclocycline hydrochloride, cisplatin, vinblastine sulfate, methoxyflurane, amphotericin B, and colchicine. Drug-induced NDI more closely resembles hereditary NDI than does NDI associated with the first group. NDI also can be seen as part of Fanconi syndrome, most likely because of the chronic hypokalemia that often occurs.
PATHOPHYSIOLOGY
AVP acts on at least two different types of receptors. The V1 (platelet/vascular/hepatic) receptors are present on nonrenal
tissues, and the V2 or renal receptor is linked to adenylate cyclase and is phosphatidylinositol-independent. In normal individuals, the antidiuretic action of AVP begins with AVP binding to the V2 receptor, which leads to a cascade of events, including the activation of adenylate cyclase and the production of cyclic adenosine monophosphate (cAMP). The final step in the pathway is an increase in specific water channels (AQP2) on the luminal surface that facilitates water transport, as shown in Figure 337.1. Normal levels of AVP are found in patients with NDI as measured by radioimmunoassay, as opposed to low levels with central DI. Extrarenal vasopressin V2-like receptors also exist that mediate vasodilation, increase plasma renin activity, and stimulate the release of factor VIIIc and von Willebrand factor. In NDI, V1 receptor responses appear to remain intact, but renal V2 receptor responses are abnormal, having no antidiuretic response to the administration of either AVP or the V2-specific agonist desmopressin acetate (DDAVP) and no increase in plasma cAMP concentration. In hereditary NDI, the V2 abnormality may be generalized (renal and extrarenal) in the X-linked form or may be limited to the kidney, as in the autosomal recessive form. The two forms can be distinguished by clinical testing. DDAVP elicits normal extrarenal responses (increased levels of factor VIII, von Willebrand factor, and tissue plasminogen activator) in patients with autosomal recessive NDI but not in patients with X-linked NDI. The two forms also can be differentiated by molecular identification of AVPR2 or AQP2 mutations.
tissues, and the V2 or renal receptor is linked to adenylate cyclase and is phosphatidylinositol-independent. In normal individuals, the antidiuretic action of AVP begins with AVP binding to the V2 receptor, which leads to a cascade of events, including the activation of adenylate cyclase and the production of cyclic adenosine monophosphate (cAMP). The final step in the pathway is an increase in specific water channels (AQP2) on the luminal surface that facilitates water transport, as shown in Figure 337.1. Normal levels of AVP are found in patients with NDI as measured by radioimmunoassay, as opposed to low levels with central DI. Extrarenal vasopressin V2-like receptors also exist that mediate vasodilation, increase plasma renin activity, and stimulate the release of factor VIIIc and von Willebrand factor. In NDI, V1 receptor responses appear to remain intact, but renal V2 receptor responses are abnormal, having no antidiuretic response to the administration of either AVP or the V2-specific agonist desmopressin acetate (DDAVP) and no increase in plasma cAMP concentration. In hereditary NDI, the V2 abnormality may be generalized (renal and extrarenal) in the X-linked form or may be limited to the kidney, as in the autosomal recessive form. The two forms can be distinguished by clinical testing. DDAVP elicits normal extrarenal responses (increased levels of factor VIII, von Willebrand factor, and tissue plasminogen activator) in patients with autosomal recessive NDI but not in patients with X-linked NDI. The two forms also can be differentiated by molecular identification of AVPR2 or AQP2 mutations.
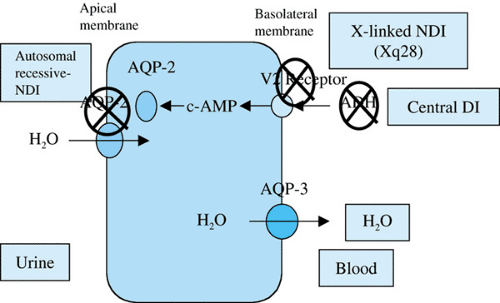 FIGURE 337.1. Schematic diagram of water transport in the principal cells of the collecting duct and genetic defects in different types of diabetes insipidus. AQP; aquaporins. |
CLINICAL MANIFESTATIONS AND COMPLICATIONS
The most common clinical manifestations of NDI are shown in Box 337.1. Polyuria is constant, even during periods of dehydration. Elevated body temperature, a consequence of the dehydration, often leads to multiple investigations that attempt to identify a possible infectious cause. The large fluid ingestion may interfere with attaining adequate caloric intake, which, along with the deleterious effects of the chronic hypernatremia, leads to physical and mental retardation. The constant need for the intake of liquids interferes with normal sleep patterns. Chronic severe constipation is another result of the constant tendency toward negative water balance that characterizes NDI. With rehydration, a dramatic reversal in the condition is seen: The signs and symptoms of dehydration disappear; the fever abates; and the vomiting, irritability, and other manifestations disappear until dehydration recurs.
BOX 337.1 Clinical Manifestations of Nephrogenic Diabetes Insipidus
Polyuria and polydipsia
Growth and developmental failure
Recurrent bouts of dehydration
Unexplained fever
Thirst
Vomiting
Constipation
Onset during infancy
Positive family history
Irritability
Laboratory Findings
Hypernatremia and hyperchloremia commonly are seen when the patient has been in negative water balance. The urinalysis usually is normal, except that the urine is inappropriately dilute, with the specific gravity being less than 1.006 and urine osmolality less than 200 mOsm/kg, despite evidence of dehydration. Small amounts of protein and a few red blood cells may be found in the urine, and the blood urea nitrogen (BUN) may be elevated during dehydration. Serum vasopressin levels are normal or high during periods of hypernatremia, distinguishing NDI from central DI. With rehydration, the sodium, chloride, BUN, and creatinine levels return to normal, and, although the urine remains dilute, the protein and red blood cells disappear. When the patient is in water balance, the glomerular filtration rate and all other renal function tests are normal, aside from the inability to conserve water. In the infant with NDI, the serum sodium level often is elevated early in the morning because of insufficient fluid intake during the night, but it may return to normal during the day with adequate fluid intake. Renal ultrasound may reveal marked dilatation of the urinary tract in NDI because of extremely high water turnover. This condition may not be apparent in very young infants but may worsen with age if control of water balance is not good. Marked dilatation of the urinary tract also may be seen in patients not identified as having NDI until later in childhood.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


