Fig. 11.1
Glucose uptake in the leg during a cycling exercise in type 2 diabetic patients and healthy controls (Modified from Martin et al. [3])
Muscle contraction also improves whole-body glucose homeostasis, since a bout of muscle contraction can enhance insulin-induced signaling. Although insulin- and muscle contraction-induced glucose transport occur via independent mechanisms, muscle contraction affects molecules involved in the insulin cascade and is required to maintain intracellular lipid balance. Excess lipid accumulation in skeletal muscle cells caused by overfeeding potently inhibits insulin signaling, while chronic exercise increases the expression of proteins involved in glucose transport and mitochondrial biogenesis and improves pancreatic insulin secretion, suggesting that muscle contraction affects organs other than skeletal muscle. Understanding the mechanistic basis for improved glucose homeostasis resulting from muscle contraction is essential for developing effective drugs and optimizing exercise-based intervention strategies for disease. This chapter summarizes the current knowledge regarding the mechanism of glucose uptake induced by muscle contraction.
11.2 Regulation of Glucose Transport by Insulin- and Muscle Contraction-Induced Signaling
Glucose uptake by skeletal muscle cells is mediated by glucose transporters (GLUTs). Two of these, GLUT1 and GLUT4, are expressed in skeletal muscle; however, the latter is responsible for the acute regulation of glucose uptake. In the basal state, vesicles containing GLUT4 are pooled in the cytosol and translocated to the sarcolemma and T-tubules; glucose transport therefore depends on the presence of GLUT4 on the muscle cell surface, which is enhanced by insulin and muscle contraction via distinct signaling pathways. The current understanding of the molecular mechanisms of insulin- and contraction-induced glucose transport in skeletal muscle cells is summarized in Fig. 11.2.
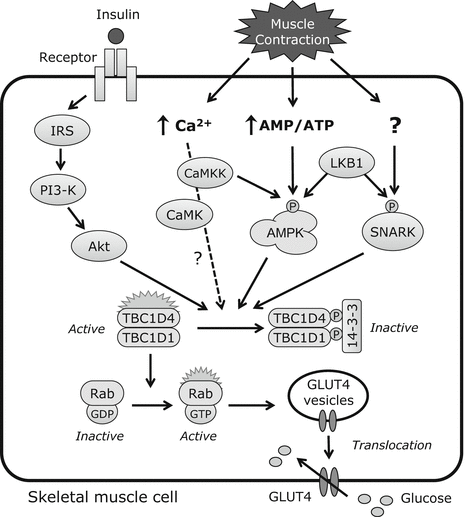
Fig. 11.2
Signaling pathways for insulin- and contraction-induced glucose uptake in skeletal muscle
11.2.1 Glucose Transport Mediated by Insulin Signaling
Insulin produced by beta cells in the pancreas is a potent stimulator of glucose transport into skeletal muscle cells. The regulation of glucose transport involving the insulin signaling pathway and the activation of various intracellular proteins has been summarized by recent review articles [5]. Briefly, the binding of insulin to its receptor triggers the activation of the associated tyrosine kinase, leading to tyrosine phosphorylation of insulin receptor substrate (IRS)-1 and activation of phosphatidylinositol 3-kinase (PI3K) followed by Akt, resulting in the translocation of GLUT4 to the muscle cell surface where glucose transport occurs via facilitative diffusion (Fig. 11.2). As described later, muscle contraction affects insulin signaling and increases insulin sensitivity.
11.2.2 Muscle Contraction Increases Glucose Transport via AMP-Activated Protein Kinase (AMPK) Activation
AMP-activated protein kinase (AMPK) is a heterotrimer composed of a catalytic α-subunit (α1 and α2) and regulatory β- (β1 and β2) and γ-subunits (γ1, γ2, and γ3). Only three combinations of these exist in skeletal muscle, namely, α1/β2/γ1, α2/β2/γ1, and α2/β2/γ3 [6]. The γ-subunit has a binding domain for ATP, ADP, and AMP and thereby acts as an energy sensor. Thr172 is the regulatory phosphorylation site of the α-subunit, and its phosphorylation is essential for catalytic activity [7, 8]. Liver kinase B1 (LKB1) was identified as an enzyme that phosphorylates Thr172 in the skeletal muscle [9] and is the major kinase regulating AMPKα: muscle-specific deletion of LKB1 abolishes AMPKα2 activity under basal and contraction-stimulated conditions [10].
A decrease in intracellular energy stores is a major trigger for contraction-induced glucose uptake. Intracellular ATP is consumed during muscle contraction, which increases AMP and ADP concentrations. This leads to the activation of AMPK, a serine/threonine kinase belonging to a family that is highly conserved among eukaryotic species and acting as a sensor of energy levels in the skeletal muscle [11]. Increases in AMP:ATP and ADP:ATP ratios cause conformational changes in the APMK complex, which induces AMPK phosphorylation by upstream kinases [12, 13].
Contraction-induced glucose uptake is mediated by AMPK activation. This was established by pharmacological experiments using the AMPK activator 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR), which was shown to stimulate glucose transport in skeletal muscle independently of the effects of insulin [14, 15]. AICAR is taken up by muscle cells and phosphorylated to yield 5-aminoimidazole-4-carboxamide ribonucleotide, a monophosphorylated derivative that mimics the effects of AMP on AMPK. The effect of AICAR treatment on glucose transport was comparable to that of muscle contraction, which was not inhibited by the PI3K inhibitor wortmannin [15]. These findings suggest that AICAR stimulates glucose transport via an insulin-independent signaling mechanism.
11.2.3 Sucrose Nonfermenting AMPK-Related Kinase (SNARK) Mediates Contraction-Induced Glucose Transport
Contraction-induced glucose uptake occurs via the LKB1/AMPK axis. The contribution of AMPK to glucose transport in skeletal muscle has been evaluated in transgenic mice that express a dominant-negative form of AMPKα in this tissue [16, 17]. As expected, AICAR- and contraction-induced AMPKα activity was completely abolished in these mice; however, AICAR- but not contraction-stimulated glucose uptake was inhibited [17]. These results suggest that AMPK regulates contraction-induced glucose transport but that other molecules are also likely to be involved.
While AMPKα2 mutant mice showed normal contraction-stimulated glucose transport [17], this was markedly reduced in muscle-specific LKB1 knockout mice lacking AMPKα2 activity [10], implying that one or more additional LKB1 substrates are involved. SNARK has been identified as a contraction-activated signal that mediates glucose transport in skeletal muscle [18]. Muscle contraction increased SNARK activity in skeletal muscles of normal mice but was abolished in LKB1 knockout mice, suggesting that SNARK is a downstream effector of LKB1 in the regulation of glucose transport [18].
11.2.4 Effect of Ca2+ Signaling on Skeletal Muscle Glucose Transport
Ca2+ released from the sarcoplasmic reticulum initiates muscle contraction and is thought to directly regulate glucose uptake. Skeletal muscle glucose uptake was increased in response to low concentrations of caffeine, a stimulator of sarcoplasmic reticulum Ca2+ release, in the absence of contractile activity and alternation of cellular energy status [19, 20]. These reports suggest that Ca2+ release is sufficient to increase glucose uptake in a signaling mechanism that is independent of AMPK. However, several studies have demonstrated that caffeine induces AMPK activation in the absence of muscle contraction [21, 22]; from this it was concluded that Ca2+ release per se does not regulate glucose transport [23, 24]. It has been proposed that increased cytosolic Ca2+ reuptake into the sarcoplasmic reticulum increases energy demand and results in AMPK activation [23].
Ca2+-sensitive signaling molecules regulates glucose transport during muscle contraction. Ca2+/calmodulin-dependent protein kinase kinases (CaMKKs) phosphorylate Thr172 of AMPK, potentially activating AMPK signaling [25, 26], and CaMKKα overexpression in mouse skeletal muscle increases AMPK Thr172 phosphorylation as well as glucose uptake [27]. Ca2+/calmodulin-dependent protein kinase (CaMK)II is also considered as a regulator of contraction-induced glucose uptake in skeletal muscles: the transfection of a CaMKII-specific inhibitory peptide into mouse skeletal muscle in vivo reduced contraction-induced glucose transport [28]. However, it has been suggested that the contribution of CaMK-mediated glucose transport is relatively low and its role is now less certain [23].
11.2.5 Downstream Effectors of Glucose Transport During Muscle Contraction
Although insulin- and contraction-induced signaling pathways in glucose uptake are distinct, both stimuli induce the translocation of GLUT4. Recent studies have shown that the insulin- and contraction-induced signaling pathways converge on some molecules in skeletal muscle. The molecules at the nexus are Tre-2/Bub2-Cdc16 (TBC)1 and domain family TBC1D1 and TBC1D4 (also known as Akt substrate of 160 kDa or AS160), which are Rab GTPase-activating proteins (GAPs) that regulate GLUT4 traffic. When TBC1D1 and TBC1D4 are phosphorylated by upstream signaling, they bind 14-3-3 protein and inhibit GAP activity, leading to the conversion of inactive GDP-loaded to active GTP-loaded Rab. The active form recruits various effectors and allows the transport and fusion of GLUT4 storage vesicles to the plasma membrane [29]. TBC1D1 and TBC1D4 exhibit GAP activity toward a number of Rab proteins [30, 31], suggesting that these participate in GLUT4 translocation via their GTPase activities.
11.3 Exercise Increases Insulin Sensitivity in Skeletal Muscles
Another important beneficial effect of muscle contraction on glucose homeostasis is the improvement of insulin sensitivity in skeletal muscle. The direct effect of muscle contraction on glucose uptake declines within a few hours, but insulin-induced glucose uptake in exercised muscle remains elevated for several hours [32–35]. This insulin sensitization by muscle contraction was first reported in an investigation of the effect of one-legged exercise on glucose uptake in response to insulin infusion [36]. The results demonstrated that glucose uptake was significantly higher in exercised than in rested legs 4 h after exercise. In support of these findings, it was shown in both humans and rodents that acute muscle contraction is associated with increased insulin sensitivity for up to 72 h after the acute effect of exercise on glucose uptake has disappeared [32, 33, 35]. Insulin-stimulated glucose transport following several hours of exercise is accompanied by enhanced GLUT4 translocation without an associated increase in GLUT4 expression or proximal insulin signaling [37, 38]. Interestingly, insulin sensitization following pre-muscle contraction is observed in insulin-resistant muscle from mice fed with a high-fat diet [37]. Therefore, insulin sensitization by muscle contraction is gaining attention for its potential application in pharmaceutics for the treatment of obesity-related diabetes.
It is unknown which molecules are responsible for insulin sensitivity after muscle contraction, but recent studies have shown that acute contraction increased phosphorylation of GAPs [32]. In experiments on isolated rat skeletal muscles, site-specific phosphorylation of TBC1D4 was increased in exercise relative to rested muscle [37, 39, 40]. It was also reported the acute exercise increased insulin-stimulated glucose transport, which was correlated with TBC1D4 phosphorylation in healthy human subjects [41, 42], whereas insulin-stimulated phosphorylation of TBC1D1 remained unaffected by pre-exercise [40, 41]. Taken together, these data suggest that muscle contraction enhances insulin sensitivity via stimulation of TBC1D4 phosphorylation.
11.4 Chronic Exercise Improves Glucose Homeostasis
11.4.1 Effect of Chronic Exercise on Glucose Transport Mediated by Insulin Signaling in Skeletal Muscle
Physical activity is known to improve glucose homeostasis in healthy and diabetic individuals. Insulin sensitivity is correlated with degree of physical activity [43]; therefore, regular exercise is a therapeutic means of improving glucose metabolism. Various studies have shown that exercise training improves glucose tolerance and insulin action in insulin-resistant individuals [44] or type 2 diabetic patients [45]. The molecular mechanism underlying enhanced glucose uptake with chronic exercise may be linked to increased expression and activity of proteins involved in glucose uptake and metabolism in skeletal muscle: chronic exercise was shown to increase GLUT4 expression and enhance glucose uptake in resting and contracting muscles [46, 47].
Exercise training improves insulin-stimulated glucose uptake by enhancing insulin signaling, which is impaired in skeletal muscles of diabetic individuals [48]. Importantly, chronic exercise increased insulin-stimulated PI3K activity—a regulatory step in glucose uptake—in skeletal muscles with a concomitant increase in PI3K activity and IRS-1 expression [40–42], suggesting that IRS-1 is likely the predominant factor responsible for exercise-induced changes in insulin signaling to PI3K in skeletal muscles.
Another mechanism for the enhancement of insulin sensitivity following chronic exercise is the upregulation of TBC1D4 phosphorylation. Actually, chronic endurance training increased TBC1D4 protein expression and its phosphorylation induced by insulin stimulation [49, 50]. Recently, Consitt et al. showed that TBC1D4 phosphorylation by insulin signaling was impaired in insulin resistance subjects, and long-term chronic endurance exercise increased whole body insulin action and reversed the impairments in TBC1D4 phosphorylation [51]. These findings suggest that increases in insulin-induced glucose uptake can be explained by altered TBC1D4 signaling.
11.4.2 Effect of Chronic Exercise on Insulin Secretion by Pancreatic Islets
Chronic exercise can affect whole-body glucose metabolism by enhancing insulin secretion by the pancreas. In general, chronic exercise is associated with a reduction in blood-insulin concentration [52]. This does not reflect a decrease in the ability of the pancreas to secrete insulin, but results from an increase in insulin sensitivity in skeletal muscles due to exercise, which reduces the amount of insulin required to maintain glucose availability.
The effect of chronic exercise on insulin secretion by the pancreas has been extensively studied, but the findings remain controversial, likely due to differences in experimental models such as the in vivo model [53, 54], isolated islets [55, 56], and single beta cell assay [57, 58] that have been used to evaluate insulin secretion. To evaluate the ability of islets to secrete insulin without the influence of an extracellular environment, our group has used isolated pancreatic islets maintained by a perfusion system to examine insulin secretion in response to high-glucose stimulation, since pancreatic beta cells sense glucose levels in the circulation and consequently secrete insulin [59]. Chronic exercise on a treadmill enhanced glucose-induced insulin secretion while pancreatic insulin level and tissue weight were unaltered [60]. It is unclear how chronic exercise (i.e., muscle contraction) improves pancreatic function, but the cross talk between the skeletal muscle and the pancreas may be explained by activities of glucagon-like peptide (GLP)-1—which is a stimulator of insulin secretion—and interleukin (IL)-6, which was shown to enhance insulin secretion by stimulating GLP-1 secretion from intestinal L and pancreatic alpha cells [61]. IL-6 is a myokine whose concentration in blood increases markedly during exercise [62]; therefore, the exercise-induced improvement of islet function may be explained by IL-6 and GLP-1 signaling.
11.5 Muscle Contraction Improves Intracellular Lipid State
11.5.1 Effect of Muscle Contraction on Lipid-Induced Insulin Resistance
Lipids are important metabolic substrates in the maintenance of skeletal muscle contractile activity. During muscle contraction, skeletal muscle cells take up fatty acids as a source of ATP. However, an imbalance between lipid supply and demand—for instance, caused by excess accumulation of intracellular lipid metabolites—leads to various metabolic disorders such as insulin resistance, type 2 diabetes, and obesity [63, 64]. Intracellular lipids are used to synthesize complex lipids including diacylglycerol, which activates serine kinases such as protein kinase C and cJun terminal kinase and subsequently inhibits insulin signaling molecules such as insulin receptor and IRS-1 [65–67]. This lipid-induced dysregulation of glucose metabolism is referred to as lipotoxicity [68].
Lipid-induced insulin resistance in muscle results from an impaired lipid oxidation capacity in mitochondria [69], which was shown to be reduced in insulin-resistant subjects relative to controls [70, 71]. An impaired capacity for oxidizing fatty acids leads to intramyocellular fat accumulation and reduced insulin sensitivity. Muscle contraction is the most effective way of reducing lipid accumulation and preventing lipid-induced insulin resistance. Chronic exercise increases the oxidative capacity of skeletal muscle by inducing the upregulation of proteins involved in mitochondrial biogenesis such as peroxisome proliferator-activated receptor-γ coactivator [72]. Endurance training was shown to enhance insulin sensitivity via increased skeletal muscle fat oxidation [73], and muscle contraction induced by electrical stimulation in the rat hindlimb increased insulin-stimulated glucose transport, which was associated with reductions in diacylglycerol and ceramide levels [74].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
 Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
 Overview
Overview
 Fracture Risk in Diabetes
Fracture Risk in Diabetes
 Body Temperature Regulation During Exercise Training
Body Temperature Regulation During Exercise Training
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


