Fig. 5.1
Effects of the AGE-RAGE axis on osteoblasts
5.5 AGE-RAGE Axis and Osteoclasts
There is some controversy about the pathological role of AGEs in osteoclast function in osteoporosis. Miyata et al. reported that when mouse unfractionated bone cells containing osteoclasts were cultured on dentin slices, AGE-modified proteins increased the number of resorption pits formed by osteoclasts [39]. Moreover, bone resorption was augmented when unfractionated bone cells were cultured on AGE-modified dentin slices [39]. They also showed that AGE-modified bone particles implanted subcutaneously in rats were resorbed to a much greater extent than nonglycated control bone particles [39]. Since AGEs did not increase the number of newly formed osteoclasts, AGEs could not promote the differentiation of osteoclasts but may activate osteoclasts or alter microenvironments favorable for bone resorption by osteoclasts. In addition, human AGE-rich cortical bone specimens were reported to increase bone resorption activities of osteoclasts [40]. These observations suggest the pathological role of AGEs in osteoclast activation in osteoporosis, which might lead to increased bone resorption and bone loss in diabetes. However, Valcourt et al. reported that when mature osteoclasts were seeded on AGE-modified bone and ivory slices, bone resorption was inhibited rather than increased due to decreased solubility of AGE-modified type 1 collagen molecules [41]. Further, they found that AGE-modified proteins inhibited osteoclastogenesis partly by blocking the osteoclastic differentiation process. So, they finally concluded that bone remodeling could be impaired in diabetes.
There is accumulating evidence that RAGE plays an important role in osteoporosis [32–47] (Fig. 5.2). Mice lacking RAGE had increased BMD and bone biomechanical strength and decreased number of osteoclasts and its bone resorptive activity in vivo [42, 43]. In vitro-differentiated RAGE-deficient osteoclasts exhibited disrupted actin ring and sealing zone structures, impaired maturation, and reduced bone resorptive activity [42]. These observations suggest that RAGE is involved in osteoclast actin reorganization, adhesion, and activation, thereby contributing to reduced bone mass in diabetes. AGEs increased mRNA levels of RAGE and receptor activator of nuclear factor-κB ligand (RANKL) in osteoblasts [44]. RANKL is an essential cytokine for osteoclastogenesis, and osteoblasts express RANKL in response to bone-resorbing factors, thus further suggesting the active participation of RAGE in osteoclastogenesis [45]. Moreover, high-mobility group box 1 (HMGB1), a nonhistone nuclear protein and one of the ligands of RAGE, has been shown to enhance RANKL-induced osteoclastogenesis both in cell culture and animal model [46].
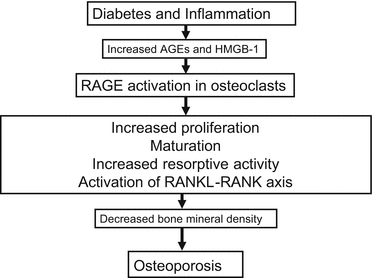
Fig. 5.2
Role of RAGE in osteoclast dysfunction
5.6 AGE-RAGE Axis and Mesenchymal Stem Cells (MSCs)
AGE-modified albumin induced generation of ROS through the interaction with RAGE and inhibited proliferation and migration of bone marrow MSCs [48]. AGEs stimulated expression and secretion of chemokines and cytokines including CC chemokine ligand (Ccl) 2, Ccl3, Ccl4, and interleukin-1β via activation of p38, which could exert the inhibitory effects on MSCs growth and migration, thereby impairing bone repair in diabetes [48]. MSCs from rats with streptozotocin-induced diabetes were more likely to become senescent, and their ability to proliferate and differentiate to the bone was reduced compared with those from control rats [49]. Glyoxal, a highly reactive dicarbonyl and one of the precursors of AGEs, which is produced by auto-oxidation of glucose, also induced senescence in bone marrow-derived telomerase-immortalized MSCs that were accompanied by increased extent of DNA breaks and AGEs accumulation [50]. Glyoxal also impaired the differentiation of MSCs determined by decreased ALP activity and reduced mineralized matrix formation as well [50]. In addition, AGEs not only inhibited the osteoblastic differentiation and growth but also induced apoptosis of mouse stromal ST2 cells [51]. Furthermore, methylglyoxal suppressed the expression of osteotrophic Wnt-targeted genes, including osteoprotegerin, a decoy receptor of RANKL via oxidative stress generation, thus causing low-turnover osteoporosis in diabetes [52]. Levels of AGEs, RAGE, ROS, and apoptosis in diabetic MSCs were increased, and extensive loss of trabecular bone in the tibiae was observed in diabetic animals [49]. AGEs inhibited the proliferation, self-renewal, and osteogenic differentiation of MSCs in vitro [49]. We also found that AGEs increased RAGE induction, ROS generation, and apoptosis and consequently inhibited mineralization and mature bone nodule formation of MSCs [53].
Up-regulation of heme oxygenase 1 (HO-1) expression and increased HO activity stimulated the differentiation of MSCs in favor of the osteoblast lineage by decreasing peroxisome proliferator-activated receptor-γ and increasing osteogenic markers such as ALP and bone morphogenetic protein-2 [54].
Taken together, these findings suggest that the AGE-RAGE-oxidative stress system could contribute to exhaustion of MSCs and loss of their differentiation potential to bone, thereby increasing the risk for osteoporosis in diabetes (Fig. 5.3).
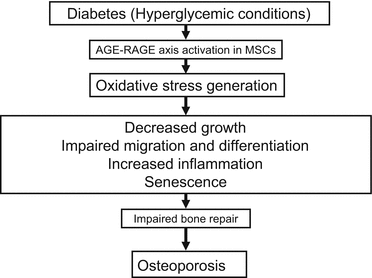
Fig. 5.3
Effects of the AGE-RAGE axis on MSCs
5.7 AGE-RAGE Axis and Bone Quality
In vitro glycation of human tibial cancellous bone cores increased the microdamage and reduced the fracture resistance [55]. AGE-modified collagen impaired lysyl oxidase enzyme-dependent physiological collagen cross-links both in primary nondifferentiated and in differentiating mouse and rat osteoblast cells via blocking the binding of collagen to discoidin domain receptor-2 [56]. AGEs accumulation in the bone also suppressed lysyl oxidase and induced bone collagen degradation in a rat model of renal osteodystrophy [57]. Further, in other animal models, despite the lack of reduction in BMD, bone mechanical properties were impaired in spontaneously diabetic WBN/Kob rats, which were coincided with decreased enzymatic cross-link formation and increased pentosidine level in the bone collagen [58]. Using a high-resolution nonlinear finite element model that incorporates cohesive elements and micro-computed tomography-based 3D meshes, Tang et al. reported that age-related increase in bone porosity and AGEs resulted in an 88 % reduction in propagation toughness [59]. Moreover, trabecular pentosidine in human vertebrae was significantly associated with whole bone strength [60]. In addition, the intensity of staining of AGEs in bone specimens of osteoporotic subjects was correlated with patient age and inversely associated with the percentage of bone surface covered with osteoblasts [61]. Vitamin C transporter expression in the type 1 diabetic mouse bone and bone marrow was suppressed which was accompanied with decreased bone formation and lower bone quality in these animals [62]. These observations suggest that mechanical integrity of the collagen network in the bone might deteriorate with diabetes and/or age due to enhanced accumulation of bone AGEs and increased oxidative stress, being involved in osteoporosis in these subjects.
5.8 Serum and Urinary Level of AGEs as a Biomarker of Osteoporosis
Serum pentosidine level was significantly increased in postmenopausal type 2 diabetic women with vertebral fractures compared with those without fractures [63]. In the same study, serum pentosidine level was also associated with the presence of vertebral fractures independent of BMD, risk factors for osteoporosis, diabetic status, and renal function [63]. Hein et al. have reported that the osteoporosis group has significantly higher serum concentrations of pentosidine and CML than healthy subjects [64]. In subgroups characterized by increased bone resorption, serum pentosidine was correlated significantly with histomorphometric marker reflecting osteoclast activity and bone resorption [64]. Shiraki et al. showed that urinary pentosidine level was correlated with time-dependent incidence of vertebral fractures in elder women who were not receiving any drug treatment for osteoporosis, whose association was totally independent of the traditional risk factors for osteoporosis [65]. When examining the relationship between baseline characteristics and incident vertebral fracture in Japanese osteoporosis patients undergoing bisphosphonate treatment, they also found that patients who developed incident vertebral fractures were older and had lower lumbar spine BMD, a higher prevalent vertebral fracture number, and higher urinary pentosidine level than patients who did not develop vertebral fractures [66]. In the Cox’s proportional hazard model, higher baseline urinary excretion level of pentosidine was one of the independent predictors of the incident vertebral fracture in these subjects [66]. Moreover, Schwartz et al. reported that elevation in urinary pentosidine level was independently associated with both increased clinical fracture incidence and vertebral fracture prevalence in elderly patients with type 2 diabetes [67].
Serum level of endogenous secretory RAGE (esRAGE)-to-pentosidine ratio in type 2 diabetic patients with vertebral fractures was significantly lower than in those without vertebral fractures [68]. Multivariate logistic regression analysis adjusted for age, serum creatinine, duration of diabetes, therapeutic agents, osteoporotic risk factors, and lumbar BMD showed that both low serum level of esRAGE and decreased esRAGE-to-pentosidine ratio were independently associated with the prevalence of vertebral fractures in patients with type 2 diabetes as well [68]. These findings suggest that serum esRAGE level and esRAGE-to-pentosidine ratio might be a more useful biomarker than BMD for assessing the risk of vertebral fractures in type 2 diabetic patients. Bone quality is more important than BMD in defining the increased risk for osteoporotic bone fractures in type 2 diabetic patients [17–19]. Furthermore, since the AGE-RAGE system plays a role in impaired bone quality in type 2 diabetic subjects [47], the authors speculated that an insufficient amount of esRAGE to counteract AGEs could intensify the binding of AGEs to RAGE and resultantly exert harmful effects on bones, thereby being involved in the increased risk of vertebral fractures in their patients. However, soluble form of RAGE (sRAGE) can be generated both from proteolytic cleavage of cell membrane surface full-length RAGE by sheddase and novel splice variants of RAGE [69, 70]. Circulating sRAGE in humans is mainly derived from the cleavage of membrane-bound RAGE, whereas esRAGE is one of the C-truncated splice isoforms of RAGE and only constitutes small part of endogenous sRAGE [69, 70]. Moreover, since interaction of RAGE with the ligands such as AGEs and HMGB1 promotes the RAGE shedding [70], it is conceivable that sRAGE level could correlate with high levels of ongoing inflammation in diabetes. Therefore, although exogenously administered high amounts of sRAGE were shown to block the harmful effects of AGEs in animals by acting as a decoy receptor [71, 72], it is questionable that esRAGE may also exert the same biological effects in humans. Serum concentration of esRAGE in humans is about 5,000 times lower than needed for the binding to and efficiently eliminating circulating AGEs [69, 73]. Taken together, decreased level of esRAGE may be associated with the prevalence of vertebral fractures in type 2 diabetes in unknown mechanisms other than working as a decoy receptor for AGEs. Since we and others have recently found that sRAGE levels are independently and inversely associated with HMGB1 level in an apparently healthy population and that sRAGE is absent and HMGB1 level is higher in diabetic RAGE−/−/apoE−/− mice [74, 75], esRAGE might protect against vertebral fractures by working as a decoy receptor for circulating HMGB1. Binding affinity of HMGB1 to RAGE is ten times higher than that of AGEs, whereas serum concentration of HMGB1 is 1,000 times less than that of AGEs [69, 74, 75], thus supporting the concept that circulating HMGB1 but not AGEs might be a molecular target for esRAGE in diabetic patients with osteoporosis.
In a cross-sectional study, 128 men and premenopausal women with type 1 diabetes, individuals with bone fractures had higher pentosidine level compared to those without fractures, while there was no significant difference of CML and esRAGE values between the two groups [76]. Moreover, multivariate logistic regression analysis revealed that the pentosidine level but not BMD was independently associated with prevalent fractures [76]. These findings suggest that osteoporotic bone fractures could result from impaired bone quality due to accumulation of pentosidine in the bone, which was unrelated with decreased esRAGE levels.
5.9 Other Biomarkers
As discussed above, oxidative stress or low levels of antioxidants are supposed to reduce BMD and cause osteoporosis in diabetic patients. An imbalance between natural antioxidative and oxidative markers was observed in patients with osteoporosis [77, 78]. Plasma total homocysteine level was higher in postmenopausal diabetes women with osteoporosis than those without osteoporosis [77]. BMD was closely correlated with homocysteine value in these subjects [78]. Homocysteine level was also shown to be inversely correlated with BMD and with both dietary intake and serum concentration of folate in Japanese type 2 diabetic patients, thus suggesting that nutritional status of folate might affect the homocysteine level, a putative risk factor for osteoporosis [78]. Furthermore, Kuyumcu et al. reported that higher serum uric acid and albumin levels were associated with a lower prevalence of osteoporosis, whereas higher homocysteine level was correlated with lower BMD and higher osteoporosis prevalence [79]. Increased uric acid levels were also associated with higher lumbar spine BMD in peri- and postmenopausal Japanese women [80].
5.10 Bisphosphonates
Bisphosphonates are a potent inhibitor of bone resorption and are one of the most widely used drugs for treatment of osteoporosis [81]. Farnesyl pyrophosphate synthase has been shown to be as a molecular target of nitrogen-containing bisphosphonates, and inhibition of posttranslational prenylation of small molecular weight G proteins is likely involved in their antiresorptive activity on osteoclasts [82, 83]. Since AGEs exert various biological actions on a variety of cells through RAGE-mediated, NADPH oxidase-induced ROS generation and subsequent NF-κB activation via Ras-MAPK pathway [84–86], it is conceivable that nitrogen-containing bisphosphonates might have pleiotropic properties by blocking farnesylation of small G proteins, which serve as lipid attachments for a variety of intracellular signaling molecules. Indeed, we have previously found that minodronate, a nitrogen-containing bisphosphonate, inhibits the AGE-induced vascular cell adhesion molecule-1 expression in endothelial cells by suppressing ROS generation via suppression of geranylgeranylation of Rac, a component of endothelial NADPH oxidase [86]. Incadronate also reverted the angiogenic activity of AGEs in endothelial cells by suppressing the RAGE-downstream signaling [85]. Moreover, AGEs significantly decreased osteoblast proliferation, ALP activity, and type 1 collagen production while increasing osteoblastic apoptosis and ROS production, all of which were completely reverted by low doses of bisphosphonates [87]. Bisphosphonates may block the deleterious actions of AGEs on osteoblastic cells via Ca(2+) influx, because the L-type calcium channel blocker, nifedipine, has been shown to inhibit the effects of bisphosphonates on AGE-exposed osteoblasts [87]. These findings could suggest a novel beneficial aspect of bisphosphonates on osteoporosis; bisphosphonates could protect against the AGE-induced bone loss partly by suppressing the RAGE-downstream signaling pathways in osteoblasts via inhibition of NADPH oxidase-mediated ROS generation.
However, it should be mentioned that 1 year of high-dose bisphosphonate therapy in dogs allowed the increased accumulation of AGEs and reduced postyield work-to-fracture of the cortical bone matrix [88]. Furthermore, pentosidine contents were increased following 3-year treatment with incadronate in dogs [89]. These observations suggest that long-term use of bisphosphonates might impair physiological bone remodeling, which could lead to an increase in nonenzymatic cross-linking in the bone, thereby altering bone matrix quality and being involved in bisphosphonate-related atypical femoral fractures [90]. AGEs disrupted the osteoblastic actin cytoskeleton and altered the cell morphology with a decrease in cell-substratum interactions, thereby causing apoptotic cell death of osteoblasts, all of which was deteriorated by the treatment with high concentration of alendronate [91].
5.11 Selective Estrogen Receptor Modulator (SERM)
Raloxifene, one of the widely used SERMs, which has estrogen-like effects on bone and “antiestrogen effects” on other tissues, has been in development for osteoporosis prevention and treatment in postmenopausal women [92]. Raloxifene has been shown to ameliorate detrimental enzymatic and nonenzymatic collagen cross-links and bone strength in rabbits with hyperhomocysteinemia [93]. ROS-activated FoxOs in early mesenchymal progenitor cells inhibited the Wnt signaling pathways, thereby impairing the osteoblastogenesis, which was prevented by estrogen [94]. Further, we have very recently found that bazedoxifene could inhibit the AGE-RAGE-induced endothelial cell damage through its antioxidative properties (unpublished data). Pullerits et al. reported that postmenopausal rheumatoid arthritis patients receiving hormone replacement therapy (estradiol plus norethisterone acetate) displayed significantly decreased serum level of sRAGE, which was associated with the elevation in serum estradiol [95]. They also found that sRAGE level at baseline was correlated with bone/cartilage turnover markers. The decrease of sRAGE level after hormone replacement therapy paralleled with diminished concentration of the markers and was correlated with an increase in total BMD in these subjects [95]. These findings further support the concept that sRAGE might be a biomarker that could reflect tissue RAGE expression in the bone and that hormone replacement therapy could exert beneficial effects on bone metabolism in postmenopausal rheumatoid arthritis patients by inhibiting the AGE-RAGE-oxidative stress axis in the bone.
5.12 Parathyroid Hormone (PTH)
Panuccio et al. reported that plasma pentosidine level was inversely related with circulating PTH and bone ALP value in hemodialysis patients [96]. The observations could suggest that AGEs accumulation may be a factor involved in low bone turnover in dialysis patients. Human PTH(1–34) treatments for 18 months were shown to increase bone volume and trabecular thickness and to decrease pentosidine level in an ovariectomized primate model [97]. Administration of human PTH(1–34) is a promising strategy for the treatment of osteoporosis; it not only increases BMD but also may improve bone quality by reducing the accumulation of AGEs in the bone of postmenopausal women.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Related posts:
 Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
 Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
 Overview
Overview
 Fracture Risk in Diabetes
Fracture Risk in Diabetes
 Body Temperature Regulation During Exercise Training
Body Temperature Regulation During Exercise Training
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


