CHAPTER 26
Management of Brain Injury Related Spasticity
Mary Alexis Iaccarino, Saurabha Bhatnagar, and Ross Zafonte
Spasticity remains among the most vexing problems after severe traumatic brain injury (TBI). Patterns of spasticity with a linked TBI can be complex and diffuse due to the multifaceted nature of TBI and its often association with polytrauma (eg, limb-based injury, burns, and fractures) (1). Spasticity can be the source of lost function, pain, contracture, and frustration. To avoid duplication with other sections in this book, this chapter focuses on the issues most applicable to those with TBI throughout the continuum of care. Due to limited literature in this population, in some cases specific reference is made to literature from parallel populations.
EPIDEMIOLOGY
The incidence of spasticity in TBI is high (2–5). In a retrospective review of rehabilitation outcomes in 122 veterans and active military personnel with combat- and noncombat-related severe brain injury, spasticity was the most common medical complication (2). In this cohort, 70% of patients developed spasticity that required treatment with oral medication, injectable, or surgical therapies. In a subgroup analysis of the same data, the likelihood of developing spasticity did not vary with the mechanism of injury (traumatic vs nontraumatic). The national guidelines regarding the management of spasticity by the Royal College of Physicians estimated that 75% of patients with severe TBI would develop spasticity requiring treatment, compared with 33.3% of patients with stroke and 60% of patients with severe multiple sclerosis (MS) (3). An evaluation of long-term outcomes by Sendroy-Terrill et al provides a more modest estimate of the incidence of spasticity after TBI. This cross-sectional study of 243 patients between 1 and 30 years postinjury found that 18% of patients reported spasticity. Furthermore, it found that the presence of spasticity had significant positive association with length of loss of consciousness (4). A 2013 study of patients with severe acquired brain injury found that 56.7% of patients suffered from spasticity in the acute care or acute rehabilitation setting (5). In the same cohort, spasticity (along with hydrocephalus, seizure, and urinary tract infection [UTI]) was found to correlate with a decreased functional status on functional independence measure (FIM) of motor and overall FIM scores at 1-year postinjury. The aforementioned highlights both the prevalence of spasticity in the severe TBI population and the importance of early treatment to prevent long-term functional impairments.
PATHOPHYSIOLOGY AND UNIQUE ASPECTS OF TBI
Any lesion of the brain or spinal cord, occurring above the alpha motor neuron, results in positive and negative symptoms that characterize the upper motor neuron syndrome (UMNS). Spasticity is a predominant positive symptom of the UMNS. Spasticity has been defined by Lance as a motor disorder characterized by a velocity-dependent increase in the tonic stretch reflexes (muscle tone) with exaggerated tendon jerks, resulting from hyperexcitability of the stretch reflex (6). This hyperexcitable state is not due to increased sensitivity or input from the muscle spindle (afferent limb of the stretch reflex) but rather due to a loss of inhibitory influences at the level of the spinal cord and in the brain. In the optimally functioning monosynaptic stretch reflex, there are several inhibitory signals that modulate the circuit including small interneurons called Renshaw cells, nonreciprocal Ib sensory afferents from the Golgi tendon organ in the muscle, and inhibitory interneurons that act on 1a afferents before their synapse with the alpha motor neuron (6,7). There are both local and descending supraspinal influences that drive these inhibitory signals. In TBI, the loss of supraspinal input to these otherwise dampening signals contributes to overexcitation of the stretch reflex and results in the clinical manifestation of spasticity.
A main supraspinal inhibitory influence is generated from the dorsal reticulospinal tract (DRT), which originates in the brainstem but receives input from the motor cortex. The DRT descends within the spinal cord and is in close proximity to the lateral corticospinal tract (LCT), a portion of the pyramidal system. Lesions affecting the DRT may also affect the LCT, thus producing a combination of pyramidal and extrapyramidal signs. Other pathways originating in the brainstem, including the excitatory vestibulospinal tract and medial reticulospinal tract, may be involved in flexor and extensor posturing seen in TBI. However, they are not thought to be as influential on spasticity (8).
It is important to note that the loss of supraspinal input occurs in brain injury (acquired and traumatic) and in spinal cord injury (SCI). However, some texts differentiate the pathophysiology of cerebral spasticity from that which occurs in spinal lesions. A review by Sheean on the pathophysiology of spasticity notes that these two general types of injuries do not necessarily produce distinct patterns or degrees of spasticity, but rather the location of the lesion within the brain or spinal cord better correlates with spasticity (9).
In TBI there is both focal and diffuse brain damage that can disrupt supraspinal signals. At the time of impact, there is a discrete surface lesion that forms as the brain contacts boney surfaces within the skull damaging areas of the gray matter and gray–white interface. The anterior temporal and frontal poles, the lateral and inferior temporal cortices, and the orbital frontal cortices are commonly injured. Concurrently, there may be a more diffuse, multifocal injury pattern within the deeper white matter preferentially affecting the corpus callosum, subfrontal white matter, and rostral brainstem. Termed diffuse axonal injury (DAI), this damage to cortical networks is caused by sheer and tensile forces and by the initiation of an excitatory cytotoxic cascade producing a neurotoxic milieu within injured areas (10). Thus, spasticity secondary to TBI is most likely not related to a discrete culprit lesion but develops when multiple foci within cortical and subcortical structures are damaged.
There often are other observed changes to active and passive movement, resting postures, and muscle tone that can be mislabeled as spasticity during a TBI clinical evaluation. Examples of this include flexor and extensor posturing, cocontraction of agonist and antagonist muscle groups, soft tissue contracture, rigidity, and spastic dystonia (Figure 26.1). Although their individual pathophysiologic mechanisms are outside the scope of this chapter, it is important to understand that they differ from spasticity (Table 26.1).

FIGURE 26.1 Severe spasticity with contracture and poor seating posture.
TABLE 26.1
MOVEMENT PATTERNS IN TBI | |
Cocontraction | The involuntary activation of antagonist muscles during voluntary contraction of agonist muscles |
Contracture | A fixed shortening of the muscle and/or surrounding soft tissues |
Flexor and extensor posturing | A hyperexcitable, nonvelocity-dependent, coordinated muscle contraction across multiple joints in response to cutaneous or nociceptive stimuli |
Spastic dystonia | Involuntary posture that may occur at rest or interfere with both passive and active voluntary movements |
Spasticity | Velocity-dependent increase in muscle tone with increased tonic stretch reflexes and exaggerated tendon jerks |
Rigidity | Increased muscle tone that is not velocity dependent |
TBI, traumatic brain injury.
ACUTE EVALUATION AND MANAGEMENT OF POST-TBI SPASTICITY
Some TBI patients will develop spasticity in the acute care setting. Each patient should be evaluated regularly for the development of spasticity so that treatment can be initiated early. Untreated spasticity is associated with increased pain, contractures, heterotopic ossification, loss of function, and skin breakdown. The development of spasticity early in the patient’s hospital course is seen most often when there is a concurrent SCI, hypoxic ischemic injury, or autonomic dysfunction.
The spasticity evaluation in the acute care setting may be challenging. Severely brain injured patients may be under sedation, on mechanical ventilation, have a concurrent infection, or metabolic derangements that will affect the assessment. Hinderer et al proposed that patients be evaluated in the following areas: clinical history, stretch examination, passive motion examination, active motion examination, and functional examination (11). Clinical history should note prior functional history, previous neurologic dysfunction, and risk factors, such as immobilization, motor dysfunction, hypoxic ischemic injury, concurrent SCI, and age. Attention should be given to the patient’s medication regimen, as several medications in the acute care setting may affect tone. Neuroparalytic agents can eliminate tone, opioids and benzodiazepines can decrease tone, and propofol (a short-acting sedative) may decrease tone. Zafonte et al also suggested using an objective measurement of spasticity, such as the Ashworth Scale, and to assess severity at different times of the day as it may change based on medication administration time, procedures, and other stimuli (1). Spasticity should also be assessed daily, as day-to-day changes may indicate serious complications not otherwise visible on other portions of the physical examination, such as hydrocephalus, new or worsening intracranial hemorrhage, cerebral edema, or infection.
Nonpharmacological and pharmacological treatments can be used to manage spasticity in the acute care setting. In general, nonpharmacological therapy is preferred in the acute TBI patient because it avoids potential central nervous system (CNS) side effects associated with many oral medications. Local chemodenervation, such as botulinum toxin (BoNT), may also be appropriate; however, BoNT should be used with caution as it may mask and impair early spontaneous motor recovery. The types of interventions and their uses are discussed in detail in the subsequent sections.
Nonpharmacological Interventions in Post-TBI Spasticity
Physical modalities are a preferred first-line treatment as they avoid potential side effects seen with pharmacotherapy. Stretching has been a mainstay of therapy in spasticity. A small study of passive stretching with flexion and extension of the elbow has been shown to be effective in reducing tone in patients with brain injury (12). An evaluation of the efficacy of stretching and passive movement of 10 vegetative and minimally conscious patients in a rehabilitation unit found improvements in caregiver ease and patients’ abilities to tolerate seating; however, there was no significant clinical improvement in joint range of motion (13). A Cochrane review of stretching in individuals with neurologic injuries showed no immediate or long-term clinical benefit (14). This review suggests that although stretching is a commonly used modality for TBI-related spasticity, the evidence of whether outcomes are affected is unclear.
Splinting and serial casting provide a period of prolonged stretching and are used in both acute and chronic TBI-related spasticity. Although the exact mechanism by which they reduce spasticity is not well understood, its efficacy is supported in the literature. Singer et al found that in patients with brain injury with spastic equinovarus deformity, serial casting for 5 weeks significantly increased muscle extensibility of the triceps surae in nine patients and decreased passive torque in eight patients. There was a median improvement in ankle dorsiflexion of 30° with knees flexed and 15° with knees extended (15).
Moseley conducted a randomized crossover study of nine patients with acute brain injury (16). Combined casting and stretching was compared with stretching alone. Torque-controlled passive range of motion (PROM) was significantly increased by a mean difference of 15.4° (P < .05) in the casted group. In a double-blind, randomized crossover study involving 15 patients with acute TBI, serial casting applied to the elbow or wrist four to six times over 1 month was compared with traditional therapy alone (17). In this study by Hill, there was a significant increase in PROM in the casted group (P = .014), improved spasticity only in the group casted first, and no effect on function in either group. A randomized controlled study by Lannin et al assessed 28 patients receiving standard physiotherapy of the upper limb within 6 months of a TBI, with 17 of the patients also receiving static palmar hand splints in the resting functional position, donned for up to 12 hours overnight (18). They found that wearing a nightly resting hand splint had no significant improvement in wrist and finger flexor muscle length, hand function, or pain compared with patients who received motor training and stretching alone. Notably there is variability in the outcomes of the aforementioned studies; this may be due to small study sample size and the heterogeneous nature of TBI.
Casting may also reduce spasticity in areas beyond the specific area being treated (Figure 26.2). Barnard et al showed a reduction in tone throughout the body after 10 days of casting the ankles of a comatose patient 10 days postinjury (19). Considerations in the use of serial casting include duration of therapy and potential treatment side effects. A retrospective case comparison study by Pohl of 105 patients with serial casting of the elbows, wrists, knees, and ankles found that duration of casting of 1 to 4 days is as effective in improving PROM as 5 to 7 days (20). However, those in this study with longer duration between casting intervals had more complications compared to those with shorter duration (29.3% vs 8.8%) including development of pressure ulcers, pain, and swelling. Although the development of pressure ulcers during serial casting was a significant finding, the same group found that appropriate serial casting for TBI patients with refractory pressure ulcers could improve positioning and facilitate pressure ulcer healing (21). Pain is another complication of splinting and casting that can have considerable downstream effects. Untreated pain can contribute to dysautonomia, agitation, and increased intracranial pressure in the acute TBI population.
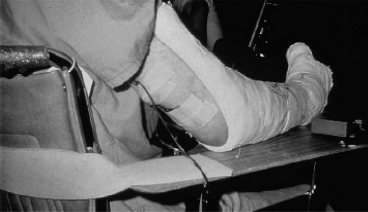
FIGURE 26.2 Electrotherapy (with antagonist stimulation) and casting for a spastic upper extremity.
There is some limited evidence for other modalities to treat spasticity in the TBI population. Both cryotherapy and electrical simulation provide short-term (hours to days) relief of increased muscle tone (22,23). Future nonpharmacological interventions for combating spasticity in this population may include noninvasive and deep brain stimulation. Human trials show some benefit in recovery of active motor function. In a double-blind, placebo-controlled, crossover study of five spastic hemiparetic ambulatory patients, repetitive transcranial magnetic stimulation (rTMS) at 1 Hz frequency was found to significantly improve walking cadence and recurvatum. There was a trend toward improvement in clinical measures of spasticity at the ankle but ultimately this was not significant (24). A larger study of 20 stroke patients with upper limb spasticity compared rTMS plus physical therapy to sham stimulation plus physical therapy in a series of 10 therapy sessions, 3 days per week. Significant improvement of at least a one-point reduction on the Modified Ashworth Scale (MAS) was seen in the rTMS group but not in the control arm (25). These novel neurostimulation approaches continue to be under investigation and may become a more viable and readily available treatment option for brain-injured individuals (26).
PHARMACOLOGICAL MANAGEMENT OF POST-TBI SPASTICITY
Tizanidine
Tizanidine is a centrally acting alpha-2 adrenergic agonist that is effective in the treatment of spasticity. It acts presynaptically and postsynaptically in the spinal cord to decrease the release of neurotransmitters aspartate and glutamate. Tizanidine may also potentiate the activity of the inhibitory neurotransmitter glycine, which inhibits pathways traveling from the rostral pons to the spine. Tizanidine is metabolized in the liver by cytochrome P450 1A2 and caution should be exercised when co-administering inhibitors of this enzyme (27).
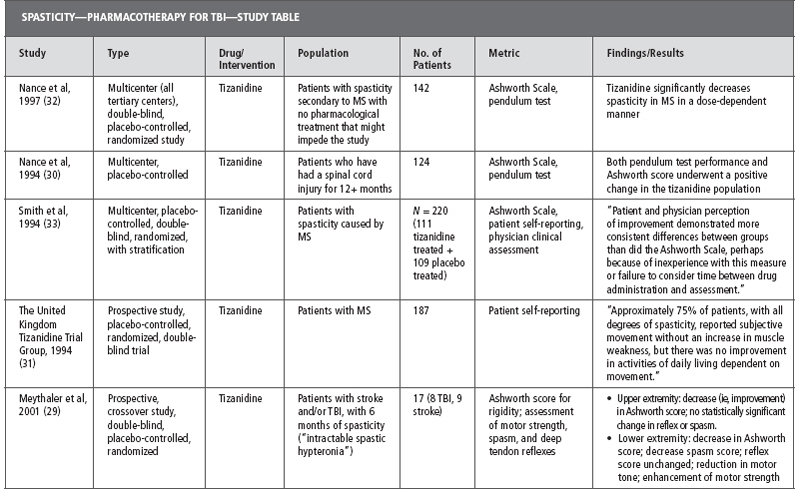
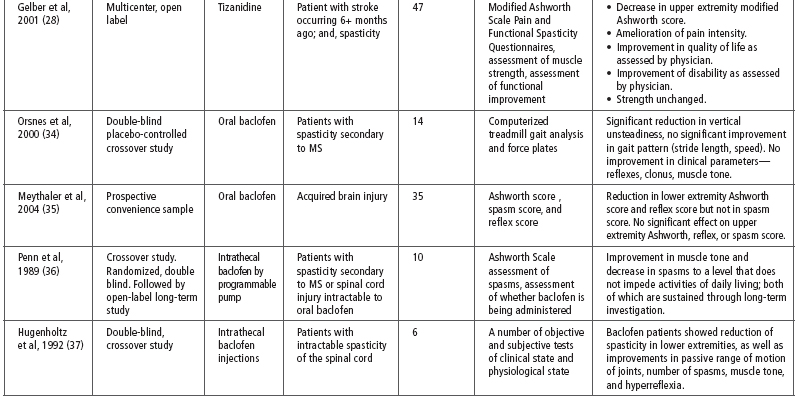
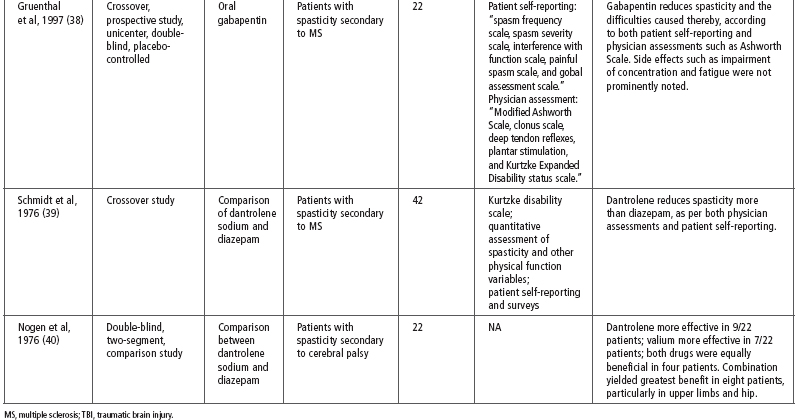
Tizanidine has been used for spasticity that is caused by a number of different diseases including SCI, MS, stroke, and TBI. Literature examining the effectiveness of tizanidine on spasticity secondary to TBI is limited and much of the evidence for its use is extrapolated from studies on other disease processes. Table 26.2 provides a summary of the evidence for the effect of tizanidine on spasticity (28–33).
Notably, one of the few studies to include patients with TBI was a small study by Meythaler et al in 2001 (29). This study included 17 patients with either TBI or stroke and refractory spastic hypertonia. Each patient was treated with tizanidine or placebo in a randomized, double-blind, placebo-controlled, crossover trial. There was a statistically significant reduction in both upper and lower extremity spasticity as measured by the MAS after 4 weeks of treatment with tizanidine. The placebo arm of this trial also saw an improved effect on tone; however, the difference seen in the active drug group was significantly more than with placebo. After 4 weeks, the average dose of tizanidine was decreased from 25.2 to 22.3 mg/d to accommodate for side effects. Tone increased during this dose reduction but there was still a statistically significant improvement in MAS from baseline. The authors concluded that tizanidine had a significant impact on severe spasticity in TBI patients and that its efficacy is dose dependent (29).
TABLE 26.2
Although evidence in the TBI population is lacking, one benefit of prescribing tizanidine for spasticity is that its side effect profile is better than some other antispasmodics, such as baclofen and diazepam. Although the structure (and possibly the mechanism of action) of tizanidine is similar to clonidine, it had a far smaller incidence of bradycardia or hypotension than clonidine when tested on animals. In addition, tizanidine does not have gamma-aminobutyric acid (GABA)-ergic or GABA-inhibitory properties, nor does it affect opioid or dopamine receptors. The major adverse effects of tizanidine include dry mouth, somnolence, asthenia, dizziness, and transient and reversible transaminitis. Although rare, visual hallucinations have been noted.
Related posts:
 Emerging Technologies in the Management of Upper Motor Neuron Syndromes
Emerging Technologies in the Management of Upper Motor Neuron Syndromes
 Effects of Noninvasive Neuromodulation in Spasticity
Effects of Noninvasive Neuromodulation in Spasticity
 Botulinum Toxin in the Treatment of Upper Limb Spasticity
Botulinum Toxin in the Treatment of Upper Limb Spasticity
 Techniques and Scales for Measuring Spastic Paresis
Techniques and Scales for Measuring Spastic Paresis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


