Malignant Bone Tumors
Murali M. Chintagumpala
The optimal management for malignant bone tumors arising in children involves a multidisciplinary approach using surgery, chemotherapy, radiation therapy, and rehabilitative therapy. Early diagnosis and prompt referral to an experienced pediatric cancer center results in a significantly improved clinical outcome in patients presenting with these aggressive tumors. The two most common malignant bone tumors in children and adolescents are osteogenic sarcoma and Ewing sarcoma.
OSTEOGENIC SARCOMA
Epidemiology
Osteogenic sarcoma is a malignant spindle-cell sarcoma of bone in which the tumor cells directly form neoplastic osteoid. Osteogenic sarcoma, or osteosarcoma, is the most common primary malignancy of bone in children. The estimated incidence is 11 cases per 1 million adolescents. The male-to-female ratio is approximately 1.5:1.0. The peak incidence occurs within the second decade, during periods of rapid growth spurts, and gradually declines thereafter.
The etiology of osteosarcoma is unknown, but several associations with underlying medical conditions have been reported. Patients who have the germinal mutation for retinoblastoma and who survive the ocular tumor have a 2,000-fold increased risk for osteosarcoma in irradiated craniofacial bones. These patients have a 500-fold increased risk for osteosarcoma at any site, regardless of prior radiation exposure. This risk appears to be linked to the expression of the retinoblastoma gene, located on chromosome 13 at band q14. Radiation-induced osteosarcoma also is being diagnosed with increased frequency in long-term survivors of childhood cancer. Pediatric cancer groups studying late effects estimate a 40-fold risk for bone cancer in survivors who have received more than 6,000 rad to the bone. The median time to onset is 10 years. An increased risk is associated with alkylating agents, proportional to cumulative doses. In older patients with Paget disease, an increased risk exists for osteosarcoma involving the affected bone. Occasional cases also have been reported in association with chondroma, osteochondromatosis, and nonossifying fibroma. Two recessive oncogenes, p53 and RB, appear to be involved either individually or in cooperation in both osteosarcoma development and progression.
Clinical Manifestations and Complications
The metaphyseal portion of the long bone is the site of predilection. Almost one-half of all new cases present with involvement in the region of the knee. In order of presentation, the most common sites are the distal femur, proximal tibia, and proximal humerus. However, any membranous bone may be involved, and even cases of extraosseous osteosarcoma have been reported.
Pain, which initially may be intermittent, and swelling of the extremity, which may evolve over several weeks, are the cardinal symptoms. Because these symptoms are nonspecific, adolescents presenting with pain in the area of the knee without a history for trauma should undergo a radiographic examination. Pathologic fractures are uncommon. However, minor trauma with disproportionate symptoms of pain may cause these patients to present for evaluation and lead to the recognition of a preexisting pathologic lesion.
Diagnosis
The diagnosis of osteosarcoma may be suspected from good-quality radiographs; tumors may appear as lytic, sclerotic, or mixed lesions. Irregular periosteal new-bone formation in the metaphyseal region may be an initial observation. In more advanced cases, cortical destruction, sclerosis, a sunburst pattern of periosteal new-bone formation, and contiguous, calcified soft tissue extensions may be noted (Fig. 311.1). Submicroscopic extension along the diaphysis can produce “skip” metastases some distance from the primary lesion.
The diagnosis is best made by incisional biopsy and permanent section. A carefully performed needle biopsy also may provide material sufficient for diagnosis. Extreme care must be taken in the biopsy of these lesions, because an incorrectly directed biopsy may produce an inadequate or misleading diagnosis or may leave a track that complicates possible consideration for limb salvage therapy. Ultimately, the biopsy track must be excised en bloc with the tumor at the time of definitive surgery. In view of the rarity of these tumors, and because of developments in the multimodal management of these patients, referral to a pediatric cancer center for definitive biopsy and diagnosis is in the patient’s best interest.
Staging of the disease should include chest radiography and computed tomography (CT) of the chest. Osteosarcoma may
spread by hematogenous routes; metastases involving the lungs or bone are detected at the time of diagnosis in 10% to 20% of the cases (Fig. 311.2). Radionuclide scans are more sensitive for detecting the foci of osseous disease distant from the primary site. Approximately 2% to 3% of all childhood or adolescent cases of osteosarcoma are multifocal. This rare type, called multifocal sclerosing osteogenic sarcoma, presents with simultaneous or synchronous metastases at multiple metaphyseal regions and has a rapidly lethal outcome.
spread by hematogenous routes; metastases involving the lungs or bone are detected at the time of diagnosis in 10% to 20% of the cases (Fig. 311.2). Radionuclide scans are more sensitive for detecting the foci of osseous disease distant from the primary site. Approximately 2% to 3% of all childhood or adolescent cases of osteosarcoma are multifocal. This rare type, called multifocal sclerosing osteogenic sarcoma, presents with simultaneous or synchronous metastases at multiple metaphyseal regions and has a rapidly lethal outcome.
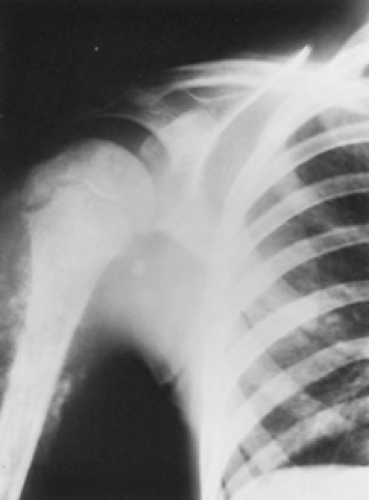 FIGURE 311.1. A large permeating lesion of the proximal right humerus, representing osteosarcoma beginning at the metaphyseal plate, with soft tissue extension and calcifications caused by osteosarcoma. |
Patients considered for limb preservation (see below) require magnetic resonance imaging (MRI) examinations of the tumor-bearing bone and occasionally angiography. MRI scans are very accurate in the assessment of intraosseous extension of tumor and are the preferred examination for patients undergoing limb salvage procedures.
Approximately 60% of adolescent cases have elevated alkaline phosphatase levels, but this does not appear to have prognostic significance.
Histologic examination permits a division of osteosarcoma into two broad categories. Patients with low-grade osteosarcomas, including juxtacortical or periosteal and low-grade central osteogenic sarcoma, have a survival rate of approximately 70% with amputation or wide local excision alone. High-grade osteosarcomas include fibroblastic, chondroblastic, osteoblastic, telangiectatic, and small cell osteosarcoma. Patients presenting with these lesions require more aggressive treatment.
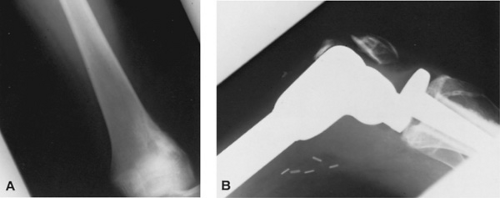 FIGURE 311.3. A: A 14-year-old girl with a small sclerotic osteosarcoma involving the distal left femur. B: Three months after receiving preoperative chemotherapy, she underwent en bloc resection and placement of an endoprosthesis, with proximal insertion into the femur and distal insertion into the tibia. |
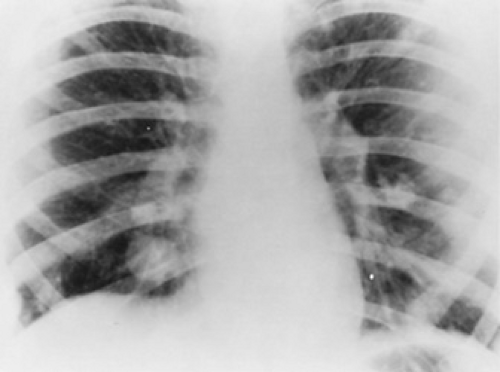 FIGURE 311.2. Chest radiograph demonstrating multiple, bilateral pulmonary nodules of metastatic osteosarcoma. |
Therapy
Before the 1970s, the prognosis for children with osteosarcoma of the extremity was dismal. Despite control of the primary tumor with amputation, distant metastases developed in most patients, and survival was approximately 20% at 5 years from diagnosis. Current multimodal treatment strategies for osteosarcoma have reversed this trend, and approximately 60% to 65% of patients with nonmetastatic disease of the extremities are surviving their disease. Both surgery and high-dose chemotherapy play a significant part in achieving this result.
Surgery has an established role in the treatment of osteosarcoma. Ablative procedures usually involve amputation through the bone above the affected bone. The general opinion is that the amputation should be several cm beyond the most proximal limits of the lesion to minimize the risk for local recurrence. Large lesions involving the proximal femur or humerus occasionally require a disarticulation procedure.
With the availability of more effective chemotherapy programs, limb salvage surgery, after en bloc tumor excision and endoprosthetic replacement, has become a viable and more frequently used alternative in patients with osteosarcoma of the extremities (Fig. 311.3). Several factors are assessed before patients are considered eligible for the limb salvage procedure, including the extent of the tumor within the medullary cavity of the involved bone, the relationship of the soft tissue component of the tumor to the major blood vessels and nerves, and evidence of joint involvement influence the type of definitive surgery to be undertaken.
Patients and the parents should be fully aware of the nature of the procedure (limb-salvage procedure or amputation), reasonable expectations for functional outcome, and estimated
risks for complications, local recurrence, and possible failure of the procedure.
risks for complications, local recurrence, and possible failure of the procedure.
Limb salvage may be performed by immediate en bloc resection or may follow a brief course of chemotherapy (neoadjuvant chemotherapy).
The potential advantages of preoperative chemotherapy are that it allows for planning and acquisition of a custom prosthesis, and the antitumor effects may enhance the safety of the surgical procedure. More important, the response of the tumor (degree of tumor necrosis) to the preoperative chemotherapy provides the single most important prognostic factor for outcome. A potential disadvantage in using preoperative chemotherapy is that the tumor is in-situ and can metastasize or the tumor can develop cells that become chemoresistant. Most pediatric cancer centers use preoperative treatment in all osteosarcoma patients. No difference in survival outcome is apparent between patients who undergo amputation at the time of diagnosis and those who had limb-salvage surgery after several courses of chemotherapy.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


