Lysosomal Storage Disorders
Rebecca S. Wappner
Lysosomes are cytoplasmic, single membrane–bound organelles that contain hydrolytic enzymes responsible for the degradation of a variety of compounds, including mucopolysaccharides, sphingolipids, and glycoproteins. The substances to be degraded are either exogenous material that has been taken into the cell by endocytosis, or endogenous material contained in cytoplasm, which has been segregated into autophagosomes. Deficient activity of a specific lysosomal acid hydrolase leads to progressive accumulation of partially degraded material, which distends the cells and disrupts cellular function. The reduced activity of an acid hydrolase may be the result of a genetic mutation at the enzyme locus that results in lowered specific activity or reduced stability of the enzyme, failure of formation of a protective protein or activator for the enzyme, or failure of formation of a recognition marker on the enzyme, which targets it for lysosomal location. Lysosomal storage of material also may result from failure of active transport of small molecules from the lysosome.
The pattern of clinical findings seen with the various disorders is related to the type of compound stored and its natural distribution in the body. All the disorders are inherited as either autosomal recessive or X-linked traits. Carrier testing and prenatal diagnosis are available for most of the disorders, but only in a limited number of experienced laboratories. An exact enzymatic and, often, molecular diagnosis is essential for accurate carrier and prenatal studies.
Current therapy mainly consists of symptomatic and supportive therapy for the patient and family. Enzyme replacement therapy (ERT) with intravenous infusions of human recombinant enzyme is presently available for Gaucher disease, Fabry disease, mucopolysaccharidosis type I (Hurler, Hurler-Scheie, and Scheie syndromes), and mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome). Clinical trials with human recombinant enzymes are currently underway for ERT for mucopolysaccharidosis type II (Hunter syndrome) and alpha-glucosidase deficiency (Pompe disease). It is anticipated that ERT for additional lysosomal storage disorders will be developed in the future. Enzyme replacement therapy decreases lysosomal storage of abnormal compounds and improves visceral and skeletal involvement, but the infused enzyme usually will not cross the blood–brain barrier to correct central nervous system involvement. Bone marrow or stem cell transplantation has also been successful in correcting many of the findings of the disorders and may be considered prior to significant central nervous system involvement. Although donor macrophages may cross the blood–brain barrier and replace brain microglial cells, it may take months or years to do so. Animal models are available that may be used for the investigation of new therapies.
For most of the disorders, the associated genes have been mapped and cloned. Heterogeneity has been noted in the molecular basis for many of the disorders and often correlates with the varying clinical presentations.
MUCOPOLYSACCHARIDOSES
The mucopolysaccharidoses are associated with lysosomal accumulation of partially degraded acid mucopolysaccharides (MPSs). MPSs, also termed glycosaminoglycans, are large molecules composed of linear repeating sulfated hexuronate or hexosamine disaccharide units attached to a protein core. MPSs normally are degraded by a series of acid hydrolases that remove the sulfate and carbohydrate residues in a stepwise manner. Deficiency of a specific hydrolase results in partial degradation of the molecules and lysosomal storage of the residual fragments.
The degradation of heparan sulfate, dermatan sulfate, keratan sulfate, or chondroitin sulfate, alone or in combination, may be involved, depending on the specific hydrolase affected. Disorders associated with heparan sulfate storage usually have central nervous system involvement and progressive mental retardation, those associated with dermatan sulfate storage have visceral and bone involvement, and those associated with keratan sulfate storage have bone involvement as their major clinical feature.
Radiography shows a distinct pattern of abnormalities termed dysostosis multiplex. The skull is enlarged and elongated (dolichocephaly) and the calvarium thickened. The sella may be J-, wooden shoe–, or boot-shaped (Fig. 389.1). The vertebral bodies in the lower thoracic and upper lumbar areas have a beaking of the anterior inferior surface caused by hypoplasia of their anterosuperior areas (Fig. 389.2). A dorsal kyphosis, or gibbus deformity, develops. The ribs are thickened, except where they join the spine, and they have an oar-shaped appearance (Fig. 389.3). The metacarpals have a proximal narrowing with distal widening, giving them a baby-bottle appearance. The distal humerus and ulna show an abnormal angulation termed a Madelung deformity (Fig. 389.4). The pelvis shows flaring of the iliac bones, shallow acetabular areas, and progressive coxa valga. The long bones become shortened, thickened, and may have signs of expansion of the medullary cavity. Hypoplasia of the odontoid process may occur. Radiography in Morquio syndrome, which is associated with keratan sulfate and chondroitin-6-sulfate storage, shows a different pattern, with a platyspondyly that resembles that seen with the spondyloepiphyseal dysplasias (Fig. 389.5).
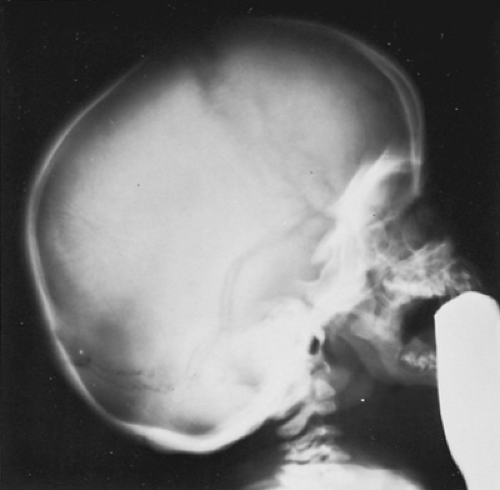 FIGURE 389.1. Lateral skull radiogram in Hurler syndrome. The skull is enlarged and elongated (dolichocephaly), with a “J” shape to the sella and thickened calvarium. |
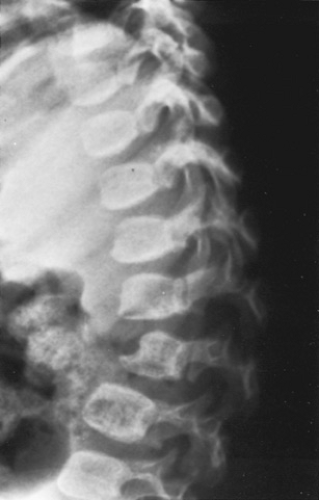 FIGURE 389.2. Lateral spine radiogram in Hurler syndrome. “Beaking” (anterior inferior projection) occurs in the lower thoracic and upper lumbar vertebral bodies from hypoplasia of the anterior superior surfaces. This results in an anterior kyphosis or “gibbus” formation. |
The age of onset, severity, and pattern of clinical and radiographic findings help to distinguish between the various types of mucopolysaccharidoses. Although urinary MPS testing may be helpful in some cases, the diagnosis is made on the basis of enzymatic testing. Demonstration of deficient activity of a specific lysosomal hydrolase may be done with serum or peripheral leukocytes for most of the disorders. Cultured skin fibroblasts may be required for others. DNA mutation analysis can confirm the disorder and may be helpful in establishing
prognosis if different forms of the disorder occur. DNA mutation analysis may also be needed for accurate carrier testing. Table 389.1 gives molecular genetic information for the MPS storage disorders.
prognosis if different forms of the disorder occur. DNA mutation analysis may also be needed for accurate carrier testing. Table 389.1 gives molecular genetic information for the MPS storage disorders.
 FIGURE 389.3. Anteroposterior chest radiogram in Hurler syndrome. The ribs are thickened, except where they join the spine, and they obscure more of the lung fields than usual. |
 FIGURE 389.4. Anteroposterior hand and wrist radiogram in Hurler syndrome. There is abnormal angulation of the distal ends of the radius and ulna and proximal narrowing of the metacarpals (baby-bottle appearance). The hands have a claw-like appearance from joint contractures and connective tissue involvement. |
Hurler Syndrome (Mucopolysaccharidosis I-H)
Hurler syndrome, the prototype for the mucopolysaccharidoses, is associated with deficient activity of alpha-L-iduronidase and excessive storage of heparan and dermatan sulfate. Hurler syndrome is inherited as an autosomal recessive trait and occurs in approximately 1 in 100,000 births.
 FIGURE 389.5. Lateral spine radiogram in Morquio syndrome. The vertebral bodies are flattened (platyspondyly). “Beaking” occurs in the lower thoracic and upper lumbar areas. |
TABLE 389.1. MUCOPOLYSACCHARIDOSES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Children with Hurler syndrome appear normal at birth. Between 6 and 12 months of age, they have the onset of gradual coarsening and prominence of facial features, with flattening of the midfacial areas and widening of the nasal bridge. Clouding of the corneas, gingival hyperplasia, and thickening of the alveolar ridge develop. Dental eruption is delayed. Deafness may occur and often is helped transiently by amplification. Respiratory involvement results from thickening of the soft tissues in the nasal and pharyngeal areas. Initially, the child may have persistent rhinorrhea or noisy breathing. Gradual upper airway obstruction results in sleep apnea and cor pulmonale. Cardiac involvement usually develops between 2 and 5 years of age and
may result in thickened valve leaflets, pseudoatheromatosis of the coronary arteries, cardiomyopathy, and congestive heart failure. Hepatosplenomegaly develops during the first year. Infrequently hypersplenism results in thrombocytopenia or pancytopenia. Umbilical and inguinal hernias often require surgical correction (Figs. 389.6 and 389.7).
may result in thickened valve leaflets, pseudoatheromatosis of the coronary arteries, cardiomyopathy, and congestive heart failure. Hepatosplenomegaly develops during the first year. Infrequently hypersplenism results in thrombocytopenia or pancytopenia. Umbilical and inguinal hernias often require surgical correction (Figs. 389.6 and 389.7).
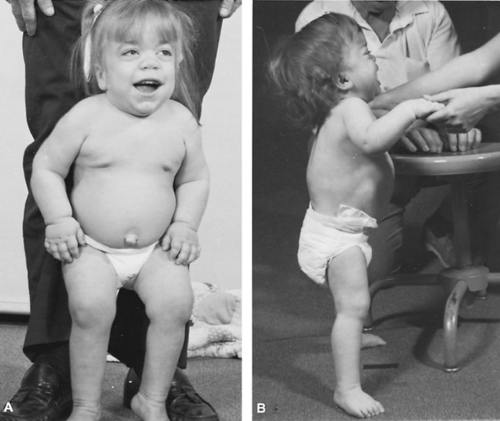 FIGURE 389.6. Children with Hurler syndrome at ages 37 months (A) and 27 months (B). Note the dolichomacrocephaly and dorsal kyphosis. |
Bone growth is delayed and there usually is minimal linear growth after 2 to 3 years of age. The gibbus deformity, a dorsolumbar kyphosis, develops during the first year and may progress. The head becomes enlarged and dolichocephalic, with prominence of the frontal areas and suture lines. Radiography shows a progression of the dysostosis multiplex. Overproduction of collagen and elastin may accompany the MPS storage and result in joint stiffness, carpal tunnel syndrome, thickening of the meninges with hydrocephalus, and decreased compliance of the thoracic cage.
Psychomotor development appears normal for the first year, remains on a plateau for 1 to 2 years, and then gradually regresses. Physical limitations are noted as a result of the joint stiffness and bone involvement. Contractures in the lower extremities lead to a “jockey stance,” and the hands become stiff and claw-like in appearance, with limited manual dexterity. Physical therapy may be prescribed, with the restriction that flexion and extension of the neck should not be done because of possible hypoplasia of the odontoid process. Adaptive equipment may be of benefit. Most children eventually become wheelchair-bound and do not live past their early teenage years. Death may occur earlier from cardiopulmonary involvement. Enzyme replacement therapy with human recombinant enzyme is currently available for patients with alpha-L-iduronidase deficiency and has shown clinical and biochemical improvement in nonneurologic manifestations of the disorder.
Hurler syndrome may be confirmed by demonstrating deficient activity of alpha-L-iduronidase in peripheral leukocytes or cultured skin fibroblasts. At least 46 mutations have been identified at the alpha-L-iduronidase locus (IDUA) that are associated with Hurler syndrome. Two common mutations, W402X and Q70X, along with one minor allele, P533R are found in over one-half of the alleles in patients from Caucasian populations. Carrier detection is available, but considerable overlap exists between carriers and noncarriers with enzymatic testing. More accurate carrier detection may be done in families of patients in whom the exact genetic mutation is known. Prenatal diagnosis is available with both chorionic villi sampling and cultured amniotic fluid cells.
Scheie Syndrome (Mucopolysaccharidosis I-S)
Scheie syndrome, formerly called MPS V, is an autosomal recessive disorder that also results from deficient activity of alpha-L-iduronidase. Most patients have mutations at the IDUA locus that allow the enzyme to retain the ability to degrade heparan sulfate. Thus, mainly dermatan sulfate is stored.
Scheie syndrome is one of the mildest forms of the mucopolysaccharidoses. Patients have normal intelligence and usually are of normal stature. Clinical symptoms begin after 5 years of age and include mild coarsening of facial features, severe clouding of the corneas, and pronounced joint involvement in the hands and feet. Aortic valvular problems are common. Carpal tunnel syndrome, degeneration of the retina, glaucoma, and deafness may occur. The disorder usually is associated with a normal or near-normal lifespan. Enzyme replacement therapy with human recombinant alpha-L-iduronidase is available.
Hurler-Scheie Syndrome (Mucopolysaccharidosis I-H/I-S)
Hurler-Scheie syndrome is a rare disorder associated with deficient activity of alpha-L-iduronidase. The clinical features are intermediate between those of Hurler and Scheie syndromes. The onset usually is during the first 2 years of life; survival has
been reported into the third decade of life. By molecular genetic analysis, some affected patients are compound heterozygotes (i.e., they have one gene for Hurler syndrome and another that allows some residual activity) or they may have other allelic mutations at the alpha-L-iduronidase locus specific for Hurler-Scheie syndrome. Enzyme replacement therapy with human recombinant alpha-L-iduronidase is available.
been reported into the third decade of life. By molecular genetic analysis, some affected patients are compound heterozygotes (i.e., they have one gene for Hurler syndrome and another that allows some residual activity) or they may have other allelic mutations at the alpha-L-iduronidase locus specific for Hurler-Scheie syndrome. Enzyme replacement therapy with human recombinant alpha-L-iduronidase is available.
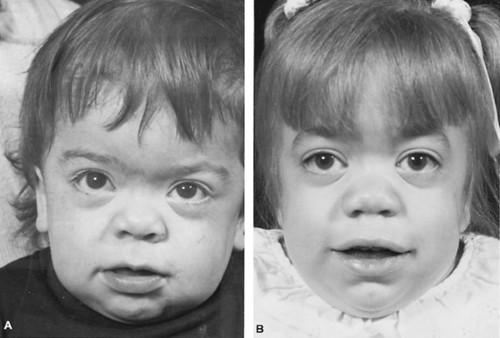 FIGURE 389.7. Face in Hurler syndrome. A: Aged 27 months. B: Aged 37 months. Prominence of facial features, increased facial hair, flat nasal bridge, and corneal clouding increase with age. |
Hunter Syndrome (Mucopolysaccharidosis II)
Hunter syndrome is an X-linked disorder associated with deficient activity of iduronate sulfatase and storage of heparan and dermatan sulfate. Both severe (type A) and mild (type B) forms exist. The clinical features of the severe form are similar to those of Hurler syndrome except that the onset is between 1 and 2 years of age, the course of the disease is somewhat slower, and no corneal clouding occurs (Fig. 389.8). Deafness is common. Skin lesions, consisting of ivory raised papules, often are noted on the upper back and on the lateral upper arms and thighs. Patients commonly survive until the second or third decades. The milder type of this disorder is comparable with Scheie syndrome, usually with normal intelligence and survival into the sixth or seventh decade. Deficient activity of iduronate sulfatase may be noted in serum, peripheral leukocytes, and cultured skin fibroblasts. Accurate carrier detection may be done only with molecular genetic techniques. Prenatal diagnosis is available using chorionic villi sampling and cultured amniotic fluid cells. Enzyme replacement therapy with human recombinant iduronate sulfatase is presently in clinical trials.
Sanfilippo Syndrome (Mucopolysaccharidosis III)
Four forms of Sanfilippo syndrome exist that are clinically indistinguishable. All are inherited as autosomal recessive traits and are associated with the storage of heparan sulfate. Type A (MPS IIIA) is associated with deficient activity of heparan N-sulfatase (sulfamidase), type B (MPS IIIB) with alpha-N-acetylglucosaminidase, type C (MPS IIIC) with acetyl CoA:alpha-glucosaminide acetyltransferase, and type D
(MPS IIID) with deficient activity of N-acetylglucosamine-6-sulfatase.
(MPS IIID) with deficient activity of N-acetylglucosamine-6-sulfatase.
 FIGURE 389.8. Brothers, ages 5 and 15 years, with Hunter syndrome. |
 FIGURE 389.9. Patient with Sanfilippo syndrome, age 6 years. Note the minimal coarsening of facial features compared with the other mucopolysaccharidoses. |
The major clinical findings are related to progressive debilitation from central nervous system involvement. Developmental delay and behavior problems usually are first noted between 2 and 4 years of age. Mild coarsening of the facial features may be noted early in the disease process (Fig. 389.9). Later, joint stiffness, hepatosplenomegaly, hernias, and radiographic findings similar to, but milder than, those seen with Hurler syndrome occur. Affected patients usually do not have corneal clouding, short stature, or cardiac involvement. Most survive into their teenage years.
The specific enzyme deficiencies may be demonstrated in peripheral leukocytes and cultured skin fibroblasts for all types. Type B also may be demonstrated in serum. Carrier detection is available for those families in whom the molecular genetic mutation is known. Prenatal diagnosis is available for all types. Bone marrow transplantation does not appear to significantly change the overall natural progression of the disorder.
Morquio Syndrome (Mucopolysaccharidosis IV)
Morquio syndrome is an autosomal recessive disorder associated with keratan sulfate and chondroitin-6-sulfate storage. Two forms of this disorder exist. Type A is associated with deficient activity of N-acetylgalactosamine-6-sulfate sulfatase (galactose-6-sulfatase) and type B with beta-galactosidase, specific for keratan sulfate. The two forms are similar in clinical findings.
The major clinical feature is skeletal involvement. Psychomotor retardation usually is not present. The onset of short stature and joint laxity occurs at approximately 1 year of age. Shortening of the trunk and neck, flaring of the ribs, prominence of the sternum (pectus carinatum), genu valgum, and enlargement and instability of the joints are noted. Mild corneal clouding and hepatosplenomegaly may be present. Enamel hypoplasia is noted in type A Morquio syndrome, but not in type B. Progressive hearing loss, either mixed or sensorineural, may require hearing aids. Cardiorespiratory problems usually are secondary to the skeletal involvement; valvular heart disease also may be present. Acute or chronic cervical myelopathy is associated with the severe hypoplasia of the odontoid process and with atlantoaxial subluxation. Posterior spinal fusion of the upper cervical spine usually is required. Both mild and severe forms of both types exist. More severely affected patients have minimal linear growth after 6 to 7 years of age and die of cardiorespiratory compromise in their third or fourth decade. Patients with milder forms have survived into their seventh decade.
Radiographic findings are evident by 2 years of age and include flattening of the vertebral bodies (platyspondyly), hypoplasia of the odontoid process, irregular metaphyses, shortening of the long bones, and findings similar to those of Hurler syndrome in the wrists and metacarpals. Keratansulfaturia is most marked early in the disease. The two types of this disorder may be confirmed by demonstration of the enzyme deficiencies in cultured skin fibroblasts. No specific therapy is available. Bone marrow transplantation has not been successful in changing the natural course of the disorder.
Maroteaux-Lamy Syndrome (Mucopolysaccharidosis VI)
Maroteaux-Lamy syndrome is an autosomal recessive disorder associated with dermatan sulfate storage and deficient activity of arylsulfatase B (N-acetylgalactosamine-4-sulfatase). Three forms of the disorder—mild, intermediate, and severe—vary in severity and age of onset. Clinical features of the severe form are similar to those of Hurler syndrome. Psychomotor retardation, however, usually is not present. Survival is possible into the third decade of life. Hydrocephalus and increased intracranial pressure may occur. Mitral and aortic insufficiency may be present. The mild form resembles Scheie syndrome except that patients are short. An intermediate type also has been reported (Figs. 389.10 and 389.11). Deficient activity of arylsulfatase B may be shown in peripheral leukocytes or cultured skin fibroblasts. Carrier detection and prenatal diagnosis are available. Bone marrow or stem cell transplantation may be considered before significant cardiac involvement. Enzyme replacement therapy is available.
Beta-Glucuronidase Deficiency (Mucopolysaccharidosis VII)
Beta-glucuronidase deficiency, also known as Sly syndrome, is an autosomal recessive disorder associated with the storage of heparan sulfate, dermatan sulfate, chondroitin-4-sulfate, and chondroitin-6-sulfate. Clinical features may be similar to Hurler syndrome in some patients and milder, without mental retardation, in others. The disorder also may present in the neonatal period with hydrops fetalis and features of a storage disorder. The enzyme deficiency may be demonstrated in peripheral leukocytes and cultured skin fibroblasts.
Hyaluronidase Deficiency (Mucopolysaccharidosis IX)
Hyaluronidase deficiency has been reported in a 14-year-old girl with short stature, periarticular soft tissue masses, and
a mildly dysmorphic facies with flattened nasal bridge. Psychomotor development and neurologic and ophthalmologic examinations were normal. Radiography of the pelvis showed nodular intraarticular soft tissue masses and acetabular erosions. Histologic studies of the masses and skin fibroblasts revealed membrane-bound storage of MPS-like material in histocytes. Plasma hyaluronidase activity was absent and plasma hyaluronan levels elevated. The disorder is inherited as an autosomal recessive trait.
a mildly dysmorphic facies with flattened nasal bridge. Psychomotor development and neurologic and ophthalmologic examinations were normal. Radiography of the pelvis showed nodular intraarticular soft tissue masses and acetabular erosions. Histologic studies of the masses and skin fibroblasts revealed membrane-bound storage of MPS-like material in histocytes. Plasma hyaluronidase activity was absent and plasma hyaluronan levels elevated. The disorder is inherited as an autosomal recessive trait.
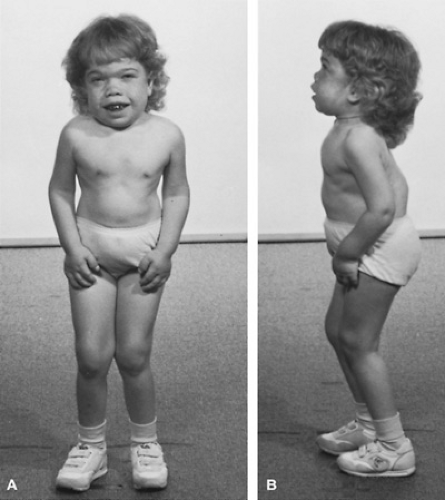 FIGURE 389.10. Patient with Maroteaux-Lamy syndrome, severe form, age 10 years. Note the coarse facial appearance, shortened thorax, hand and joint contractures, and “gibbus” formation of the thoracic spine. |
SPHINGOLIPIDOSES
The sphingolipidoses are associated with lysosomal accumulation of glycosphingolipids, gangliosides, and sphingomyelin. Faulty degradation of the molecules results from deficient activity of a lysosomal acid hydrolase or a missing sphingolipid activator protein needed for enzyme-lipid stabilization and interaction.
Ceramide, the basic structure for these molecules, is composed of sphingosine, to which a long-chain fatty acid, usually C16, has been attached at the amino group (Fig. 389.12). Attachment of neutral carbohydrate groups in an oligosaccharide chain occurs at the hydroxyl group of the sphingosine. The attachment of a glucosyl residue in beta linkage as the first neutral sugar leads to a glucosylceramide (glucocerebroside) series of glycosphingolipids. Attachment of a galactosyl residue leads to a galactosylceramide (galactocerebroside) series. The neutral sugars of the oligosaccharide side chain may be in alpha or beta linkage and are derived from glucose, galactose, N-acetyl-galactosamine, or N-acetyl-glucosamine. If the first neutral sugar is a sulfated galactosyl, the compound is called a sulfatide. If sialic acid, or N-acetyl-neuraminic acid, is attached to the neutral sugars, the structure is termed a ganglioside.
 FIGURE 389.11. Structure of sphingomyelin. A, sphingoid (sphingosine); B, fatty acid (stearoyl); C, ceramide; D, phosphoryl choline. |
 FIGURE 389.12. Eyes in adult-onset Maroteaux-Lamy syndrome. Note the corneal clouding. |
Current nomenclature of the gangliosides, according to the Svennerholm classification, is determined by the number of sialic acid residues that are attached to the oligosaccharide chain (M = mono, D = di, T = tri) and by the number (5 minus n) of neutral sugars in the chain. For example, GM1 ganglioside would have one sialic acid residue and four (5 minus 1) neutral sugars in the oligosaccharide chain attached to the ceramide. Degradation of glycosphingolipids involves stepwise removal of the neutral sugars, sulfate, and sialic acid by a series of lysosomal hydrolases (Fig. 389.13). Table 389.2 gives molecular genetic information for the sphingolipidoses.
GM1 Gangliosidosis
GM1 gangliosidosis is inherited as an autosomal recessive trait and associated with acid beta-galactosidase deficiency. Storage of GM1 ganglioside, GA1 ganglioside, keratan sulfate, and other glycoproteins and oligosaccharides that contain beta-galactoside residues occurs in the brain, viscera, bones, and other tissues of affected patients. Three major types of the disorder vary in clinical severity and age of onset, corresponding to different mutations at the beta-galactosidase gene locus.
Type 1 (infantile form or generalized gangliosidosis) is associated with a complete lack of acid beta-galactosidase activity (Fig. 389.14). Symptoms begin at or shortly after birth and include severe progressive central nervous system degeneration, coarse facial features, corneal clouding, macroglossia, gingival hyperplasia, hepatosplenomegaly, hernias, joint stiffness, dorsal kyphosis, and edema of the extremities. Cherry-red macular spots may be seen in one-half of the patients. Radiography shows dysostosis multiplex. Foamy histiocytes may be noted in bone marrow and visceral organs. Death usually occurs by 2 years of age. Carrier detection and prenatal diagnosis are available.
 FIGURE 389.13. Examples of sphingolipid degradation. A, GM1 ganglioside; B, asialoganglioside; C, sulfatide; 1, GM1-beta-galactosidase (GM1 gangliosidosis); 2, beta-N-acetylgalactosaminidase, beta-hexosaminidase A and B (GM2 gangliosidoses); 3, GM3-alpha-neuraminidase; 4, alpha-N-acetylgalactosaminidase, alpha-galactosidase B (Schindler disease); 5, alpha-galactosidase A (Fabry disease); 6, ceramide-lactoside beta-galactosidase; 7, beta-glucosidase (Gaucher disease); 8, arylsulfatase A (metachromatic leukodystrophy); 9, beta-galactosidase (Krabbe disease); Cer, ceramide; Gal, galactose; GalNAc, N-acetylgalactosamine; Glc, glucose; NANA, N-acetylneuraminic acid, sialic acid. |
Type 2 (late infantile or juvenile GM1 gangliosidosis) is a milder disorder with less severe symptoms of MPS storage. The
onset of ataxia between 1 and 2 years of age is followed by progressive mental and motor deterioration, spasticity, seizures, and blindness. Death occurs between 3 and 10 years of age. There usually is no coarsening of the facial features, hepatosplenomegaly, corneal clouding, or macular changes. Radiography may show mild changes of dysostosis multiplex.
onset of ataxia between 1 and 2 years of age is followed by progressive mental and motor deterioration, spasticity, seizures, and blindness. Death occurs between 3 and 10 years of age. There usually is no coarsening of the facial features, hepatosplenomegaly, corneal clouding, or macular changes. Radiography may show mild changes of dysostosis multiplex.
Type 3 (adult chronic GM1 gangliosidosis), the mildest type of the disorder, presents with dysarthria, gait disturbance, and a slowly progressive dystonia as early as 4 years of age, but more often in the teenage years. Intellectual involvement and bone changes, if present, are mild. Survival into early adulthood is possible. This type of the disorder occurs most commonly in Japan.
GM2 Gangliosidoses
The GM2 gangliosidoses are a group of autosomal recessive disorders associated with cerebral degeneration secondary to lysosomal storage of GM2 ganglioside and related glycosphingolipids. The disorders are associated with deficient activity of the beta-hexoaminidases or the GM2 activator protein.
Beta-hexosaminidase has two subunits, alpha and beta, which are the products of two separate genetic loci, HEXA and HEXB, located on chromosomes 15 and 5, respectively. The isoenzymes of beta-hexosaminidase are composed of different combinations of these subunits. Hexosaminidase A is composed of an alpha and a beta subunit, whereas hexosaminidase B contains two beta subunits. Hexosaminidase A usually accounts for 55% to 70% of the total hexosaminidase-specific activity, whereas hexosaminidase B accounts for 30% to 45%. Genetic mutations at the alpha subunit gene locus lead to hexosaminidase A deficiency and Tay-Sachs disease. Mutations at the beta subunit gene locus lead to deficiency of both hexosaminidase A and B and result in Sandhoff disease. A GM2
activator protein, encoded by the GM2A locus on chromosome 5, is needed for stabilization of the GM2 ganglioside-hexosaminidase A complex. At least 93 mutations at the HEXA locus, 26 at the HEXB locus, and 4 at the GM2A locus have been characterized to date.
activator protein, encoded by the GM2A locus on chromosome 5, is needed for stabilization of the GM2 ganglioside-hexosaminidase A complex. At least 93 mutations at the HEXA locus, 26 at the HEXB locus, and 4 at the GM2A locus have been characterized to date.
 FIGURE 389.14. Patient with GM1 gangliosidosis. This child has a mildly coarse facial appearance, facial edema, and an enlarged abdomen from hepatosplenomegaly. An early “gibbus” formation was noted in the thoracic spine. |
TABLE 389.2. SPHINGOLIPIDOSES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


