Leukodystrophies
Alan K. Percy
The leukodystrophies are a group of progressive inherited neurodegenerative disorders affecting myelin formation in the central and (in some instances) peripheral nervous systems. As such, they should be distinguished from disorders characterized by demyelination, such as multiple sclerosis, inflammatory disorders, or toxic processes. The leukodystrophies can be categorized as dysmyelinating (abnormal myelin formation), hypomyelinating (failure of myelin formation), or vacuolating (dissolution of myelin). The leukodystrophies are associated mainly with infants and children, but adult variants are recognized with increasing frequency. These disorders are recessive conditions involving both autosomal and X-linked inheritance. Typically, the clinical pattern of the leukodystrophies is characterized by the loss of previously acquired capabilities, whether motor or cognitive, signifying a degenerative disease. In infants, this condition reflects loss of acquired developmental milestones, whereas during childhood or adolescence, changes in behavior or school performance are more typical.
When the degenerative process occurs in infancy or early childhood, often white matter or myelin dysfunction can be distinguished from gray matter or neuronal dysfunction on the basis of the clinical appearance of the disorder. Children with disorders of white matter display a loss of acquired motor capabilities (e.g., ambulation or hand use), hypotonia, or ataxia or signs of corticospinal tract involvement, such as spasticity, hyperreflexia, or extensor plantar responses. Children with disorders of gray matter demonstrate intellectual or cognitive deficits, such as loss of language or communication skills, seizures, and blindness. This formulation may be an effective guide to conducting clinical and laboratory assessments early in
the disease process. Global nervous system involvement limits the effectiveness of this approach during later stages.
the disease process. Global nervous system involvement limits the effectiveness of this approach during later stages.
During infancy, the various types of leukodystrophy must be differentiated from an emerging static encephalopathy. In the presence of rapid, clear-cut regression, making this determination is straightforward. Subtle regression or a plateau in development, however, presents a greater diagnostic challenge. In older children, consideration must be given to such demyelinating processes as multiple sclerosis, acute disseminated encephalomyelitis, or polyneuropathy (e.g., Guillain-Barré syndrome). In addition, other conditions must be excluded, including nutritional deficiency, a toxic encephalopathy (from heavy-metal or drug intoxication with such drugs as neuroleptics, hypnotics, or anticonvulsants), a neoplasm of the central nervous system (CNS), an immunopathologic condition (systemic lupus erythematosus), or a chronic infectious process (human immunodeficiency virus, acquired immunodeficiency syndrome, or subacute sclerosing panencephalitis).
As a group, the leukodystrophies have an incidence of one in 50,000, based on recent data from Germany. This incidence figure should be regarded as a minimum number because underdiagnosis is likely. Efforts to provide useful screening tools have focused on cranial magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) analyses. In particular, cranial MRI has increased the recognition and elucidation of an emerging group of leukodystrophies, such as vanishing white matter disease (see Section, Vanishing White Matter Disease). Cranial MRI also is an effective mechanism for monitoring the progression of disease and assessing the efficacy of treatment during the conduct of clinical trials. Effective treatment presently is limited for those entities described in the following sections.
Metachromatic leukodystrophy and globoid cell leukodystrophy (Krabbe disease) are autosomal recessive disorders of sphingolipid metabolism. The sphingolipids are a group of unique lipid compounds that are important constituents of biologic membranes. The tissue distribution of the individual sphingolipids differs dramatically. Some sphingolipids are prominent within neural elements, and others are present in non-neural tissues. The sphingolipids sulfatide and galactosylceramide are critical components of myelin in both the central and peripheral nervous systems, whereas the group of sphingolipids called gangliosides are associated particularly with neurons and their axons and dendrites. (Disorders of ganglioside metabolism are described in Chapter 389, Lysosomal Storage Disorders.)
Adrenoleukodystrophy is an X-linked disorder of very long-chain fatty acid (VLCFA) metabolism. Canavan disease, an autosomal recessive disorder, is caused by abnormal acetylaspartate metabolism. Pelizaeus-Merzbacher disease, an X-linked disorder, is caused by duplications or point mutations of the proteolipid protein (PLP) gene. Alexander disease, an autosomal recessive disorder, results from mutations in the gene for glial fibrillary acidic protein. Vanishing white matter disease is caused by mutations in genes encoding the eIF2B translation factor.
Effective therapy for these disorders is limited. Bone marrow transplantation has been performed with variable success. Gene therapy strategies are being investigated. Dietary therapy with erucic acid (Lorenzo oil) has been employed without clear benefit in clinically affected children, but it may be beneficial in delaying onset in the presymptomatic stage. Symptomatic therapy should be used as indicated to treat infections, seizures, and other medical problems. Affected families should be provided with appropriate support, as the stress associated with caring for such children can be overwhelming.
The prenatal diagnosis of many of these disorders is feasible. Analysis of amniotic fluid cells or chorionic villus samples can provide an accurate assessment of the fetus. Population-based screening currently is not realistic. As such, prenatal diagnosis is practical only in families with a known risk for having affected offspring. Despite the identification of specific mutations in these disorders, in many instances, a determination of the relevant enzyme or metabolite remains the preferred diagnostic modality.
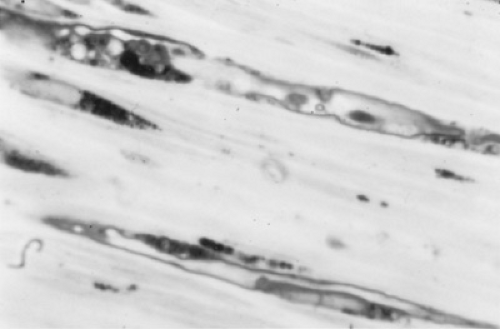 FIGURE 411.1. Light micrograph of peripheral nerve in longitudinal section from a patient with metachromatic leukodystrophy, showing lipid inclusion material within the Schwann cell cytoplasm and in the interstitial space. |
METACHROMATIC LEUKODYSTROPHY (SULFATIDE LIPIDOSIS)
Metachromatic leukodystrophy is an autosomal recessive disorder with an incidence of one per 40,000 people. The biochemical basis is the inability to degrade the sphingolipid, sulfatide, or galactosylceramide-3 sulfate (Figs. 411.1 and 411.2). Sulfatide, along with galactosylceramide, is an important constituent of central and peripheral nervous system myelin. Metachromatic leukodystrophy results from a deficiency of sulfatide sulfatase (arylsulfatase A), the lysosomal enzyme responsible for the degradation of sulfatide. This deficiency causes an accumulation of sulfatide in both neural and non-neural (especially kidney and gallbladder) tissues, where it can be detected as metachromatic granules. Both central and peripheral myelin
are abnormal. Neuropathology consists of the widespread loss of myelin and oligodendroglia in the brain and segmental demyelination of the peripheral nerves.
are abnormal. Neuropathology consists of the widespread loss of myelin and oligodendroglia in the brain and segmental demyelination of the peripheral nerves.
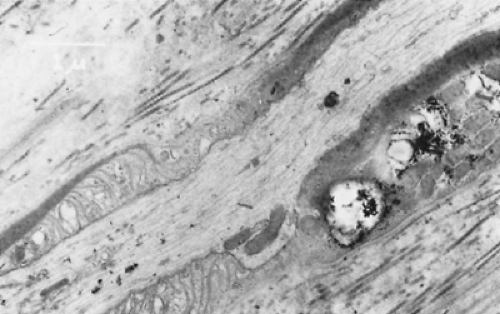 FIGURE 411.2. Electron micrograph of peripheral nerve in longitudinal section from a patient with metachromatic leukodystrophy, showing densely staining inclusion material within the Schwann cell cytoplasm. |
TABLE 411.1. CHARACTERISTICS OF SULFATIDE LIPIDOSES | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
Forms of the Disease
Metachromatic leukodystrophy occurs in three principal forms: late infantile (the most common), juvenile, and adult (Table 411.1). The distinction of separate phenotypes is arbitrary, however, as this variability reflects the continuum of phenotypes rather than separate entities. As specific mutations are identified, the genotype-phenotype correlations become clearer and the continuum of clinical involvement becomes more evident. The late infantile form appears in affected children between ages 1 and 2 years, after they have experienced normal early development, often including ambulation. Early signs are regression of motor skills, gait difficulties, ataxia, hypotonia, and extensor plantar responses. Muscle stretch reflexes are diminished or absent as a result of peripheral nerve involvement. Optic atrophy is prominent early. The disorder progresses fairly rapidly, with loss of motor and mental capabilities and ultimately loss of meaningful contact with the environment. Death usually occurs by the time the child is age 6 years.
Juvenile metachromatic leukodystrophy may represent two patterns of disease. An early juvenile form begins in children between ages 4 and 8 years, with the development of gait disturbance and intellectual decline. Ataxia and upper motor neuron signs are prominent. Muscle stretch reflexes are increased initially but subsequently are lost. Clinical progression is less rapid than that in the late infantile form. Often, extrapyramidal signs and seizures appear, and death commonly occurs within 6 years of onset.
Alternately, juvenile metachromatic leukodystrophy can begin in affected children between ages 6 and 16 years as personality and behavior changes and declining school performance. Seizures are common occurrences, and motor dysfunction eventually ensues. The progression of this form is slower still, and survival is possible into late adolescence or early adulthood.
The adult form of metachromatic dystrophy presents as dementia or psychosis as early as age 16 years or as late as age 60 years. Often, declining school or work performance is the first sign of the disease. Motor dysfunction is an inevitable development, but its progression may be very slow.
Multiple sulfatase deficiency combines features of the late infantile form of metachromatic leukodystrophy, steroid sulfatase deficiency, and the mucopolysaccharidoses (see Table 411.1). Mucopolysaccharide storage is suggested by the presence of coarse facial features, ichthyosis, hepatosplenomegaly, and skeletal abnormalities. Tissue accumulation of sulfatides, sulfated steroids, and mucopolysaccharides reflects the pervasive deficiency of multiple sulfatase activities.
Diagnosis and Therapy
The diagnosis of metachromatic leukodystrophy is accomplished by careful clinical assessment and the performance of appropriate laboratory studies. The diagnosis is suggested by a history of regression and progressive deterioration of motor function, and signs of gait difficulties with weakness, hypotonia, hyporeflexia, and extensor plantar responses in the early forms, or behavioral and motor disability in older children or adolescents. Nerve conduction velocities are reduced as a reflection of peripheral nerve involvement. The CSF protein level is increased. Cranial computed tomography (CT) or MRI scans reveal symmetric white matter lesions in the early forms of the disease and may demonstrate cortical atrophy in the later forms. Decreased activity of the enzyme arylsulfatase A, preferably in leukocytes or skin fibroblast cultures, establishes the diagnosis. The availability of biochemical analysis has eliminated the role of peripheral nerve biopsy, which typically will reveal metachromatic granules.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


