Fig. 1
Overview of image modalities for intravital microscopy. (a) comparison of characteristic image properties (i.e., axial and lateral resolution, acquisition speed, depth penetration multichannel detection, and motion artifact vulnerability) of the most widely used microscope modalities for intravital microscopy in atherosclerotic research. The color coded areas (wide field, yellow; spinning disk, blue; CLSM, green; TPLSM, orange) represent the differences and advantages of each modality for intravital imaging. (b) Properties of novel image modalities with potential future application for intravital imaging of atherosclerosis; multiscale opto-acoustics (purple) and the nanoscopic techniques STED and RESOLFT (red)
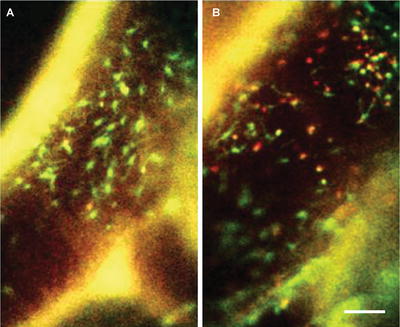
Fig. 2
Dual-channel conventional fluorescence intravital microscopy of carotid arteries. Apoe −/− mice were transplanted with Cx 3 cr1 gfp /wt Ccr2 rfp /wt bone marrow. Local treatment of carotid artery with saline (a) or TNF (b) for 4 h. Scale bar represents 75 μm. Arterial wall is visible in both channels (yellow ; autofluorescence)
An alternative approach is confocal microscopy (Fig. 1a), a modality that offers optical sectioning by blocking out of focus light, and thus enables 3D imaging. There are basically two types of confocal microscopy that can be applied for intravital microscopy offering fast multichannel 3D imaging. The most often applied intravital confocal modality is spinning disk microscopy [29] where a so-called Nipkow disk generates the confocal effect with multiple foci [30]. Spinning disk confocal microscopy is a fast modality and, dependent on the system layout and settings, the optical resolution is mainly improved in depth (maximum axial resolution ≈ 5 μm). Moreover, two or more spectral channels are typically available for simultaneous detection of multiple targets. Yet, the imaging of structures deep in or at the luminal border of the large arteries is still restricted due to the limited penetration depth. Moreover the increased cross talk between multiple foci that may occur potentially reduces the signal-to-noise ratio especially when imaging at depth in tissues. A second confocal approach is the use of a single focus, fast confocal laser scanning microscopy (CLSM) which makes use of pinhole(s) to block out of focus light and a scanhead enabling xy-scanning at resonant speeds. A state-of-the-art commercially available resonant scanned CLSM enables image acquisition at a rate of >30 Hz [31]. Combined with a piezo-driven z-positioner it is possible to obtain 3D xyz-stacks of the arterial wall at high speeds (typically 0.1–0–3 Hz) [32] (Fig. 3). Combined with powerful excitation laser sources and several sensitive emitted light detectors such as photo multipliers or (hybrid) diodes, these systems allow intravital imaging of circulation as well as the processes and structures in the tunica adventitia and media of large arteries [33]. Yet, even powerful CLSM systems lack the penetration depth that is required for studying cell recruitment at the luminal border of large arteries [33]. Fast two-photon laser scanning microscopy (TPLSM) offers a similar system layout as fast CLSM or makes use of multiplication of the excitation laser beam (1–64 beams) [34, 35], to enable high-speed acquisition without a negative impact on the overall image quality, size of the field of view, nor the image resolution. However, unlike conventional fluorescence microscopy, TPLSM makes use of a different excitation process where near simultaneous absorption of two photons of approximately half the energy (and thus double the wavelength) causes fluorescent molecules to move from the ground state to a higher energy state (in most cases similar fluorophores as those used with conventional fluorescence) [36]. The subsequent decay causes the molecule to fall back to its ground state while emitting fluorescent light of a slightly lower energy (i.e., longer wavelength). To achieve two-photon excitation, a less scattering, pulsed, near-infrared laser source is used which increases light penetration into the artery while reducing the overall energy load on the tissue. Moreover, since the two-photon excitation and the resulting fluorescence decay of (intrinsic) fluorophores only occurs in the focal spot, all the detected fluorescence signal can be used for image reconstruction. As a result, there is no need for pinhole(s) and as such the detection efficiency and the signal-to-noise ratio are further improved, enabling imaging at greater depth in the arteries or detection of slightly weaker or more vulnerable fluorescent targets [33].

Fig. 3
Luminal tracking of myeloid cells in the stabilized carotid artery. TPLSM obtained 4D datasets (XYZT) of the stabilized carotid artery of Lysmegfp/egfp Apoe−/− mice presented over time as 2D extended depth of field pictures using maximum intensity projection (a), cell tracking (b) or as 3D reconstructions with (b, c) or without (d) isosurface rendering. Arrows indicate the direction of flow, green are myeloid cells, blue is collagen derived second harmonics generation in the tunica adventitia, red is 150 kDa Tritc-Dextran in the circulating blood representing the lumen. Scale bar = 25 μm. Figure was adapted from Chevre et al. [32]
Microscopes offering high spatial and axial resolution have in common that they suffer severely from motion artifacts that are present in living specimen. When studying murine cardiovascular samples such as the large arteries, the motion disturbances are a consequence of the cardiac (6–9 Hz) and respiratory (2–4 Hz) cycles (depending on applied anesthesia) [37]. The cardiac cycle is the most prominent disturbance due to its higher frequency but causes a somewhat smaller tissue drift; only the artery moves as a consequence of the blood pressure wave. The respiratory cycle motion causes the whole thorax to move and results in a more pronounced focus drift but at a lower frequency. As a result of the tissue motion, microscopic imaging suffers from both intra-frame and inter-frame motion artifacts. The intra-frame motion artifacts only occur with laser scanning microscopes that utilize pixel by pixel image construction where a gradient in time difference between the first scanned pixel in the left top corner of the image matrix and the last scanned pixel at the right bottom corner is present. As such, the tissue in focus may vary within the time required for acquisition of one single image, causing a shift in tissue in focus and thus a deterioration of the tissue in view due to the actual motion and loss of focus [24, 32]. The tissue motion also causes a difference of the tissue in focus between subsequent images. As a result, accurate tracking of objects over time or generation of 3-dimensional reconstructions (either in depth or over time) is almost impossible without any motion suppressing aid since subsequent optical sections do not contain the same tissue over time or are not only in axially (z) shifted but also spatially (xy). In order to reduce the impact of motion on imaging, optimization of the microscope hardware (fast acquisition and high sensitivity) combined with adaptions to the experimental layout are required.
There are several strategies to (partially) overcome both artifacts: (a) utilize the cardiac and respiratory cycle to manually guide the imaging by timing of the TPLSM acquisition in a time window where the motion will be limited; (b) record the moving tissue randomly, apply extensive post-processing of the images series to detect comparable tissue representations over time; (c) trigger the acquisition by using the cardiac and respiratory signals (triggered acquisition); and (d) stabilize and (partially) isolate the artery from the surrounding tissue with specifically designed tools to suppress the impact of motion. These strategies are discussed below:
1.
Manual timing of the image acquisition between the tissue movements by means of timed recording is a logical step to (partially) overcome in vivo motion artifacts. Provided the image acquisition frequency is higher than the movement frequencies, it is possible to record a single image without intra-image artifacts. Timing of image acquisition has been performed for imaging of the lung using intravital laser scanning microscopy [38, 39]. By mechanically controlling the respiration, a stable respiration motion frequency is obtained that allows image acquisition in between the respiration frequency without intra-image artifacts (only when acquisition frequency is higher than motion frequency). When respiratory motion frequencies are unstable or when the motion is also caused by the cardiac cycle, as is the case in the large arteries, it is impossible to block the impact of motion by simple timing of acquisition in the respiration-free time window.
2.
When randomly recording a vast amount of single xy-images over time at 10–30 Hz, the resultant image datasets mainly consists of motion disturbed optical slices. Such datasets can be subjected to extensive post-processing of the images series in order to detect comparable tissue representations over time. In most cases, the (partially) stable images have a rhythmic appearance (dependent on acquisition frequency and type of motion disturbances) which is beneficial for the selection process. Additionally, non-moving structures or cells within the selected images have to be compared using overlays, edge detection, or co-localization protocols. The matching images may then be combined and used for evaluation of myeloid cell recruitment [5], vasa vasorum [40, 41], or biological structures such as presence of luminal neutrophil extracellular traps in atherosclerotic arteries [42]. The post-processing method can be performed manually but without automatic (structure recognition based) image processing protocols, it is very time consuming and tedious work with an unclear outcome; it is unclear whether the analyzed time series contain enough stable and comparable images to reconstruct cellular processes over time. Hence, there is a need for (development of) automatic processing protocols to automatically analyze in vivo datasets. To generate multidimensional microscopic datasets it is also required to have knowledge on the motion frequencies and tissue shifts which further complicates the post-processing procedures even more. Moreover, the time course of the resulting movie is not stable because of its dependency on the tissue in the focal plane.
3.
By measuring both the respiratory and cardiac cycle signals using a balloon pressure and an ECG or blood pressure probe it is possible to determine a time gate where the motion of both cycles is limited. Image acquisition of each single image in the time gate of the combined cycles (by coupling the single-trigger pulse directly into the microscope hardware in order to avoid software jitter) results in a strong reduction of both intra- and inter-frame artifacts [24]. Moreover, the acquired images of both cardiac and respiratory triggered microscopy are less influenced by small irregularities that occur in both motion frequencies due to the possibility to apply trigger thresholds or trigger delays for each signal separately as well as the overall trigger output signal to start the image acquisition. As a result, double triggered acquisition holds potential for multidimensional microscopy, especially when combined with fast (>10 Hz) spinning disk or laser scanning microscopes in order to further suppress intra-frame artifacts. A disadvantage of double-triggered acquisition is the reduction of overall imaging rates since the relatively small time gates of each individual cycle must be (partially) overlapping in order to achieve image stability. In practice, an overall acquisition frequency of 0.5–1.0 Hz is maximum achievable [24], which limits its applicability for studying fast biological processes.
4.
Besides the rather non physiological procedures of physical suppression of the tissue motion either by adapting the surgical preparation method in order to temporary stoppage of the respiration [43] or blood flow [44], more subtle stabilization of arteries can be beneficial to reduce motion impact. Attempts to stabilize arteries by embedding the exposed carotid artery in stiffened material such as low percentage agarose in buffer, have up till now failed since they do not suppress the motion disturbances sufficiently (own observations). Chevre et al. [32] used a surgical layout similarly as applied by Yu et al. [44] but stabilized the carotid artery without the dramatic alterations to the blood flow by making use of a thin steel plate to carefully lift the artery up from the surrounding tissue, and part of a cover slip to gently squeeze the artery to further reduce motion while creating a imaging window. As a result, conventional intravital microscopy with a high magnification and numerical aperture objective could be applied to study several classes of leukocytes, including monocytes, lymphocytes, and platelets, using a combination of fluorescent reporter genes and fluorescently labeled antibodies that were injected intravenously before imaging. The authors used the artery-stabilizing protocol also in combination with multichannel fast TPLSM. For application of TPLSM the method required additional triggering to further suppress the impact of the respiratory cycle on imaging. Nevertheless the combination of physical stabilization of the sample with respiration controlled image acquisition enabled recording of multidimensional datasets with relatively high frequency (0.1–0.3 Hz per z-stack) for prolonged time (>30 min) and for the first time demonstrated the feasibility of 3D tracking of cells and their function in atherosclerotic prone arteries in vivo [45].
As is evident, the described techniques have their advantages and disadvantages, and ultimately the optimal technique offers the best balance between acquisition speed, sensitivity, and (3D) resolution. The latter will depend on the research question at hand as well as the availability of the various techniques. Quantification of cell adhesion and cell-arterial wall interactions are best performed using conventional fluorescence microscopy due to its ease of use. Studies focused on tissue structure would greatly profit from the most stable imaging layout with a double triggering protocol and using modalities with optical sectioning. Fast processes however, such as multidimensional tracking of cells in the arterial system, may benefit more from artery stabilization (combined with respiratory triggering) since triggering-based techniques would result in too slow acquisition for proper assessment of real-time dynamics. In the near future, continuous imaging combined with automated post processing may enable more researchers to apply advanced microscopic techniques in strongly motional samples and indirectly obtain an improved image quality without the need for trigger adapted setups.
3 Strategies to Label Target Structures
Early intravital microcopy studies analyzing myeloid cell extravasation used bright field microscopy. Such techniques were typically employed in tissues that were thin enough to allow white light transmittance through the sample. However, as this is not possible for optical imaging of large arteries in vivo fluorescence microscopy is widely used benefiting from an ever-growing array of available fluorophores that can be used to label cells and tissue structures. These fluorophores can roughly be divided into two categories: endogenous reporters, i.e., fluorescent proteins expressed by cells of interest (Table 1); and exogenous probes, i.e., chemicals that interact with cellular or tissue components. Irrespective of the nature of the probe, fluorophores for in vivo use are ideally nontoxic, photostable, with sufficient quantum yields to generate a signal detectable through living tissues.
Table 1
List of mouse strains with fluorescent phagocyte subsets
Strain name | Fluorescent cell type | Reference |
|---|---|---|
c–fms-gfp | GFP in mononuclear phagocytes | [96] |
Cx 3 cr1-gfp | GFP expression correlates with CX3CR1 transcription. Inflammatory monocytes are GFPlo, resident monocytes are GFPhi, NK cell subsets, microglia, and DCs are GFPint | [46] |
Lysm-gfp | GFP expression correlates with the transcription of Lysozyme M. Circulating neutrophils are GFPhi, while monocytes are GFPlo | [97] |
MhcII-gfp | DCs are GFPhi, B cells are GFPint, and macrophages are GFPlo | [98] |
Cx3cr1-gfpCcr2-rfp | Inflammatory monocytes are GFPloRFP+, resident monocytes are GFPhiRFP− | [48] |
Ly6G-gfp-cre | This mouse strain is currently being developed by Dr. Matthias Gunzer (University of Duisburg-Essen, Germany); reported to carry tdTomato and Cre in neutrophils, only | Unpublished |
hCD68–gfp | This mouse strain is currently being characterised by Dr. David R. Greaves (University of Oxford, UK); the eGFP transgene under control of the human CD68 promoter is expressed in circulating monocytes and resident and inflammatory macrophages | Unpublished |
The use of transgenic mice expressing fluorescent proteins in a cell-specific fashion is an elegant method for specific cell labeling and is widely accepted to track individual leukocyte subsets. A variety of strains has been created to allow for tracking of myeloid cell populations in vivo (Table 1). However, care has to be taken when using mice where the gene encoding for a fluorescent protein has been knocked into the locus of a gene involved in recruitment. This is the case for Cx3cr1-egfp mice or Ccr2-rfp mice, where homozygous mice lack these receptors and thus recruitment of monocyte subsets may be affected [46, 47].
The generation of blue/yellow-shifted GFP variants (blue, cyan, and yellow fluorescent proteins), together with the discovery- and mutation-based development of red fluorescent proteins has expanded the palette of colors available and has allowed the development of more complex reporter strategies. This development has led to the establishment of reporter mice for multiple cell types or cell subsets. An example herein is the Cx 3 cr1 gfp/wt Ccr2 rfp/wt mouse allowing for in vivo tracking of monocyte subsets [48]. Apart from simultaneous visualization of multiple cell types, the extended palette of fluorescent proteins available has led to some very elegant applications from labelling different phases of the cell cycle to different clones of cells within a tissue. For instance, the FUCCI reporter system allows real-time visualization of cell cycle progression in living cells. Recently, a new type of FUCCI reporter strategy, based on the use of mCherry and Venus fluorescent proteins, has allowed similar studies to be performed within live, cultured mouse embryos [49]. Although the FUCCI technology has not been used in adult tissues (not to mention atherosclerosis research) this strategy could be important to study the dynamics of cell proliferation within atherosclerotic lesions. Several additional strategies for genetic labelling of cells are available. However, most of these have only been tested in cancer research and their use for optical imaging of atherosclerosis is at its infancy. Such approaches include confetti transgenic mice [50] for studying tissue dynamics, Forster resonance energy transfer (FRET)-based strategies to study signaling events [51, 52], and the use of photoswitchable or photoconvertible fluorescent proteins allowing for labeling of specific cells to be identified through multiple imaging sessions [53, 54]. On the other hand, the clear advantages of fluorescent protein-encoding mouse strains over adoptive transfer approaches is that the target cells are not subjected to ex vivo handling and that the fluorescent tags do not become diluted or fade in dividing cells.
Dyes present a variety of different challenges and limitations in comparison with endogenously expressed fluorescent proteins, ranging from higher cytotoxicity to rapid dilution upon cell proliferation. However, they is often chosen because of their ease of use and the wide array of colours they provide. Moreover, a good staining strategy may be applied in any transgenic mouse model and as such offers great flexibility in usage since there is no need to generate novel transgenic mouse lines with multiple reporters. Cytoplasmic and nuclear dyes are, in principle, ideal for in vivo single-cell tracking because they should provide the most uniform staining; however, they tend to present higher cytotoxicity and are rarely used for intravital microscopy. Carboxyfluorescein succinimidyl ester (CFSE) and carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) are amine-reactive probes that penetrate cells, where they are metabolized to amine-reactive chemicals, which then covalently bind to cytosolic components. High concentrations of CFSE have been associated with severe toxicity [55]; however, once carefully optimized, CFSE labelling enables in vivo cell tracking over a long period of time because it is very efficiently retained within the cytoplasm [56]. Amine-reactive dyes, available with a wide spectral range and in far red-shifted variants, overcome the limitations of CFSE/CFDA-SE and have been successfully used for confocal intravital microscopy (IVM) experiments.
Dyes with high affinity for double-stranded DNA provide a bright nuclear signal, and therefore should be ideal for tracking cells within densely clustered populations and in deep tissues, but have been consistently reported to have a number of limitations when applied to in vivo single-cell resolution imaging. In fact, DNA-binding dyes can interfere with fundamental biological processes, including DNA replication and transcription, and can induce DNA damage. For example, Hoechst 33342 has been reported to inhibit the proliferation of mammalian cells at high concentrations [56] and DRAQ5 was shown to interfere with DNA-binding proteins such as histones, DNA repair, replication, and transcription factors as well as essential cellular enzymes resulting in the inhibition of cellular functions [57]. Hoechst 33342 has the advantage of absorbing light in the UV range, excitable by two-photon microscopy, and is resistant to quenching. For these reasons, it has therefore been widely used to study leukocyte migration, a process uncoupled from DNA dynamics and therefore not affected by nuclear dyes. Moreover, Hoechst 33342 has been successfully used to visualize apoptosis in vivo [58]. The vital cell DNA binding dyes from the SYTO series are very stable and available with a variety of spectral properties. SYTO13 (green spectral range) and SYTO41 (blue spectral range) have been used for (in vivo) imaging of large arteries and are an alternative for Hoechst 33342 [24]. Propidium iodide also binds to DNA by intercalating between the bases with no sequence preference. As it is cell impermeable it is a good dye for in vivo detection of cell death or for detection of extracellular DNA [42].
Lipophilic, carbocyanine fluorescent tracking dyes, whose aliphatic portion binds to the cell membrane lipid bilayer, have been widely used by the scientific community and seem to have lower cytotoxicity than cytoplasmic and nuclear dyes [59]. PKH lipophilic dyes such as PKH2, PKH67 (green), PKH3 and PKH26 (red) have been extensively used for tracking leukocytes in vivo. These dyes stain the whole plasma membrane of cells through lateral diffusion, and also spread to intracellular organelles as a consequence of membrane turnover. More recently, another series of carbocyanine lipophilic dyes (DiO, DiI, DiD and DiR, with green, red, far-red, and near-infrared emission, respectively) have become increasingly popular and are now routinely used for in vivo tracking and imaging studies [60, 61]. They all follow the same labeling principle and cover a large spectral range, thus facilitating tracking of multiple cell populations simultaneously through multicolor imaging. Some of these dyes, for example DiR, are highly photostable, and all are easy and rapid to apply. Most importantly, despite the fact that they heavily intercalate within the lipid bilayer of cell membranes, they have not been reported to cause serious cytotoxicity at concentrations that provide strong, uniform staining.
In recent years, the use of mice expressing fluorescent proteins and fluorescent dyes has been complemented by the application of fluorescent antibodies towards myeloid cell subsets. The injection of antibodies to Gr1 and CD115 or F4/80 allows for labeling neutrophils or monocytes [6, 62] independently of the genetic background of the mice. However, one has to bear in mind that some antibodies may affect leukocyte behavior. In the case of antibodies to Gr1 it has been shown that higher doses of this antibody induce depletion of neutrophils, so that low concentrations have to be used to avoid such depletion [63]. In addition, antibodies may induce signaling events that impair cell function. In this context it has recently been reported that injection of low doses of anti-Ly6G antibody to label neutrophils disturbs their recruitment [64]. An alternative antibody-based approach is the conjugation of antibodies to fluorescent microbeads. Endothelial immobilization of such beads allows for detection and even quantification of cell adhesion molecule (CAM) expression, whereof the amount of immobilized beads correlates with luminal CAM expression [65] or luminally deposited chemotactic molecules (Fig. 4) [5, 7, 8]. The main challenges raised by the use of intact antibodies for in vivo labeling of cells and tissue structures are tissue penetration (i.e., the efficiency of labelling is likely to rapidly decrease with distance from vasculature), potential nonspecific signal and aberrant immune cell responses. The last two problems, both due to the antibodies binding and activating cellular Fc receptors, can be alleviated by removal of the Fc part of the antibodies [66]. An alternative to antibodies may be provided by aptamers, short nucleotide or peptide sequences that can recognize specific protein domains. Because aptamers are small and change conformation upon binding of their target, they can be engineered so that specific binding releases a quencher or generates a FRET pair, thus reducing nonspecific staining [67]. So far, aptamers have been used to label mesenchymal stem and progenitor cells ex vivo and to visualize them in mouse bone marrow following transplantation [67].
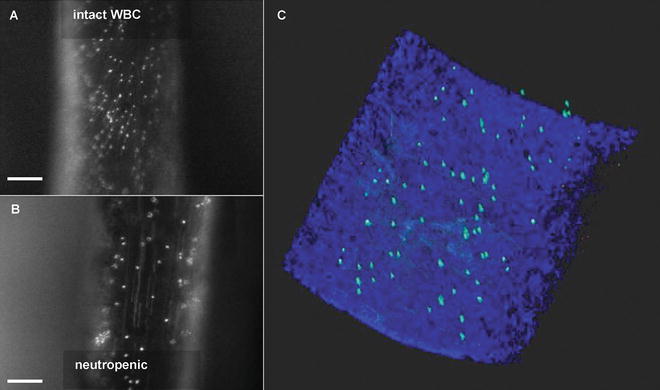
Fig. 4
Detection of luminally immobilized molecules. Conventional fluorescence intravital microscopy of anti-CRAMP fluorescent bead complex deposition in the injured carotid artery of Apoe −/− mice with intact white blood cell counts (a) and neutropenia (b). Directly after intravital microscopy, luminal presence was confirmed in the arteries ex vivo (c) using the 3-dimensional properties of TPLSM combined with intrinsic fluorescence derived from the arterial wall. Adapted from [26]
4 Current and Future Developments
In vivo imaging of cell biology processes has been increasingly rewarding, thanks to the constant improvement of fluorophores, both proteins and chemical, and to parallel technological advances. Recent developments in these fields are already providing indications on the future avenues of IVM studies. For example, fluorescent proteins emitting in the near-infrared region (such as Neptune and eqFP650/670) and the infrared region (such as iRFP) are becoming popular because they are more efficiently excited and detected in living tissues, where light scattering, autofluorescence and absorption are highest at lower wavelengths. These proteins have so far been successfully used for whole-body imaging experiments in mice [68–70]. Furthermore, near-infrared dyes are ideal candidates to enable multimodal optical imaging, for example a combination of fluorescence molecular tomography (FMT) modality offering a macroscopic view combined with CLSM or conventional fluorescence microscopy to study the same target at a subcellular resolution level. While red fluorophores are currently imaged through confocal microscopy, optical parametric oscillator (OPO) technology is making red and far-red fluorophores amenable to TPLSM [71], the ideal IVM modality as it reduces phototoxicity and increases contrast and resolution especially at higher tissue depths. Future generations of OPO technology are more flexible because of a continuous power output at the full tuning range (from 800 to 1300 nm) and as such are more easy to use and will further contribute to make the family of near-infrared fluorophores more easily detectable with multiphoton excitation. Moreover, the optical hardware will be further improved to enhance the sensitivity of near-infrared emission. Alternatively, the development of fluorophores that are excitable by short wavelengths but emit far red and near-infrared signals provides an ideal labelling strategy for IVM. Keima as well as LSS-mKate1 and LSS-mKate2 are proteins engineered with a large Stokes shift, e.g., to absorb similarly to CFP but emit above 600 nm, and other fluorescent proteins could be similarly engineered to allow two-photon excitation [72].
Related posts:
 Analysis of Gene and Protein Expression in Atherosclerotic Mouse Aorta by Western Blot and Quantitative Real-Time PCR
Tandem Stenosis to Induce Atherosclerotic Plaque Instability in the Mouse
Analysis of Gene and Protein Expression in Atherosclerotic Mouse Aorta by Western Blot and Quantitative Real-Time PCR
Tandem Stenosis to Induce Atherosclerotic Plaque Instability in the Mouse
 Quantification of Cellular Proliferation in Mouse Atherosclerotic Lesions
Quantification of Cellular Proliferation in Mouse Atherosclerotic Lesions
 Intravital Microscopy in the Cremaster Muscle Microcirculation for Endothelial Dysfunction Studies
Intravital Microscopy in the Cremaster Muscle Microcirculation for Endothelial Dysfunction Studies
 Isolation and Culture of Aortic Smooth Muscle Cells and In Vitro Calcification Assay
Isolation and Culture of Aortic Smooth Muscle Cells and In Vitro Calcification Assay
 Quantification of Apoptosis in Mouse Atherosclerotic Lesions
Quantification of Apoptosis in Mouse Atherosclerotic Lesions
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


