, Jacob Stephen2, Miranda C. A. Kramer3, Elena R. Ladich1, Frank D. Kolodgie1 and Renu Virmani1
(1)
CVPath Institute, Inc., Gaithersburg, MD, USA
(2)
Department of Cardiology/Medicine, William Beaumont Army Medical Center, Uniformed Services University of the Health Sciences, El Paso, TX, USA
(3)
Department of Cardiology, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands
Abstract
Pathology of high risk atherosclerotic plaque provides the basis for understanding the imaging and treatment of atherosclerosis. The earliest vascular change described microscopically are adaptive intimal thickening and fatty streaks, whereas pathologic intimal thickening are the first of the progressive plaques subtypes. Fibroatheromas are characterized by an acellular necrotic core, accumulated cellular debris and cholesterol monohydrate, and a lack of extracellular matrix. The development of the necrotic core is believed to originate from apoptotic macrophages. Thinning of the fibrous cap leads to plaques vulnerable to rupture, or thin-cap fibroatheromas. Overlying thrombosis can arise from one of several mechanisms including ruptures, erosion, or calcified nodules. Calcium within atherosclerosis is a common imaging target which increases with lesion progression and is present in greatest frequent in healed plaque ruptures and fibrous plaques. Thin cap fibroatheromas most frequently contain speckled calcification but may show heavily calcified areas or an absence of calcification. which is not very useful in diagnosing these lesions by calcium-based imaging. Coronary lesions with thrombi in the absence of rupture (erosions) exclusively show stippled or no calcification. Rupture in the absence of calcification is rare. In contrast, diffuse calcification is almost always associated with healed ruptures.
Keywords
PathologyAtherosclerosisVulnerable plaqueThrombosisCalcificationBackground
Atherosclerosis is a complex disease with a multi-factorial etiology related to inheritance, and traditional and nontraditional risk factors. Despite major medical advances in the treatment of atherosclerosis, approximately two thirds of patients remain refractory to statins, one of the most successful agents targeted for the prevention of myocardial infarction. The lack of a more substantial treatment effect underscores the limitations of lipid lowering monotherapy and emphasizes the complexity of the disease and requirement for multi-targeting of other critical processes. Moreover, despite the rapid progress in newer and refined imaging modalities that can image atherosclerosis, the inability to completely characterize atherosclerotic lesions in individual patients presents another important issue. Therefore, advancing the field is contingent on a better understanding of the morphologic characteristics of high-risk plaques in living patients, who harbor the capability of producing symptomatic events. Insights into how critical elements influence lesions stability primarily involve:
macrophage foam cells
necrotic core size
fibrous cap thickness
neoangiogenesis/hemorrhage
Any advancement(s) towards designing therapies targeted at plaque stabilization will likely require the identification putative pharmacologic agents and clinical refinements to validate and monitor treatment effects, potentially with arterial imaging techniques. Finally, a better understanding of the temporal relationship between active and healing lesions is needed to recognize the natural changes in plaque composition caused by silent or symptomatic events.
While intensive research has lead to a few breakthroughs in preventative therapies such as lipid lowering, most mechanistic insights are yet to be translated into new treatments. This limitation partly exists since the precise causes(s) of lesion progression from asymptomatic stable fibroatheromas into high-risk plaques for rupture (thin cap fibroatheroma or vulnerable plaque) are incompletely understood.
Natural progression of atherosclerotic plaque in humans
In animal models of atherosclerosis, hyperlipidemia-induced macrophage infiltration of the intima, constitutes one of the earliest pathologic changes [1], which can be reversed if the dietary cholesterol intake is reduced and/or circulating cholesterol is decreased by pharmacologic means. As an endpoint, however, advanced animal lesions have limited resemblance to man since findings of luminal thrombi attributed to rupture are rare [2, 3]. Nonetheless, early atherosclerosis in humans is recognized in all populations irrespective of the presence of risk factors as early as the first decade [4].
Atherosclerotic lesions have been extensively studied at autopsy where specific plaque morphologies have been assigned to categories established by the American Heart Association consensus group, lead by Dr. Stary in the mid-1990s [5, 6]. Our laboratory subsequently modified this classification as the cause of coronary thrombosis is not exclusive to rupture as implied by the AHA, but alternatively includes erosion and eruptive nodular calcification. In addition, the thin-cap fibroatheroma, the assumed precursor lesion to rupture (vulnerable plaque), was also introduced since this definition is also missing in the AHA classification.
Atherosclerotic Plaque Morphologies
Non-progressive Atherosclerosis (Fig. 1.1a–f)
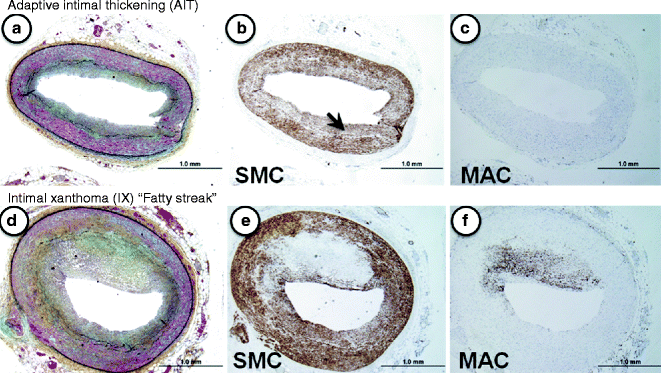
Fig. 1.1
(a–f) Lesion morphologies consistent with non-progressive atherosclerosis. (a–c) Adaptive intimal thickening (AIT). Normal coronary vessel with a thin smooth muscle-rich neointima (b, α-SMC actin immunostaining, arrow). Note the absence of lesional macrophages (MACs, c). (d–f) Intimal xanthoma (IX) or so-called “fatty streak.” Serial sections of the same eccentric plaque show few α-actin positive SMCs (d), while CD68-positive macrophages are very prominent (f)
The earliest vascular change described microscopically is adaptive intimal thickening (AHA Type I), which is found in at least 30 % of neonates at birth. The next category represents fatty streaks (AHA Type II), which are characterized by non-raised lesions consisting of intimal macrophages with intra- and extracellular lipid deposits. These lesions tend to regress in certain locations, e.g., thoracic aorta and the mid right coronary artery [7].
Progressive Atherosclerosis (Fig. 1.2a–f)
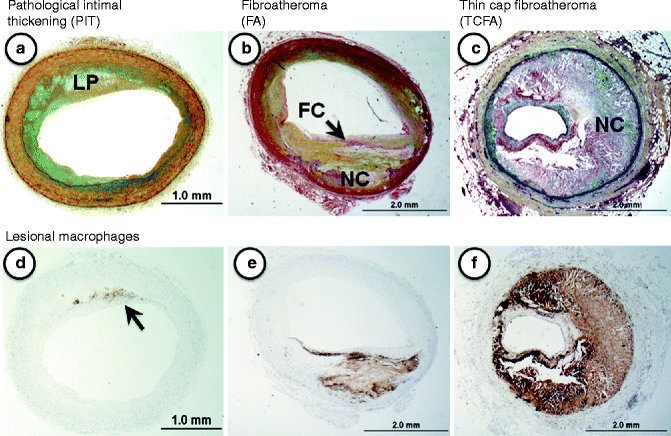
Fig. 1.2
(a–f) Lesion morphologies consistent with progressive atherosclerosis: (a–c) Movat pentachrome staining; (d–f) CD68 immunostaining = macrophages). (a) Pathologic intimal thickening (PIT) is characterized by a non-flow limiting smooth muscle cell-rich lesion with an acellular lipid pool (LP) containing proteoglycan. (b) Fibroatheroma (FA) represents a lesions with a relatively thick fibrous cap (FC) overlying an area of necrosis or necrotic core (NC). These lesions are also generally non-flow limiting. (c) Thin-cap fibroatheroma (TCFA), shows a relatively large necrotic core with a thin fibrous cap typically infiltrated by macrophages and T-lymphocytes. The TCFA or “vulnerable plaque” is a know precursor to rupture. (d–f) These show the varying distribution of macrophages in progressive plaques. Note in (d) (PIT) the macrophages (arrow) are located near the luminal surface outside the area of the lipid pool, which is a distinguishing feature of this plaque
Our laboratory recognizes pathologic intimal thickening, also known as the intermediate (AHA Type III) lesion, as the first of the progressive plaques [8, 9]. Lipid pools rich in proteoglycans (hyaluronan and versican) located near the medial wall, in areas that generally lack smooth muscle cells, define this lesion type. The luminal surface, however, is mostly rich in smooth muscle cells and often accompanied by infiltrating macrophage foam cells [10]. The precise origin of the “lipid pool” is debatable, although studies suggest that a loss of smooth muscle cells promoted by apoptosis may be involved as basement membranes remnants can be identified by periodic acid Schiff (PAS) staining. Another characteristic of these lesions is microcalcification, which is prominently seen with anionic stains such as the von Kossa’s stain [9].
The first of the advanced lesions are considered fibroatheromas (AHA Type IV), which are characterized by an acellular necrotic core, accumulated cellular debris and cholesterol monohydrate, and a lack of extracellular matrix [5, 8]. During the evolution towards a fibroatheromatous lesion an overlying layer of fibrous tissue (fibrous cap) becomes identifiably distinct from the circumscribed area of necrotic core. The fibrous cap has a critical role in harboring the contents of the necrotic core, and its integrity is one of the defining influences on plaque stability (AHA Type V) [5]. The development of the necrotic core is believed to originate from apoptotic macrophages.
The extent of fibrous cap thinning along with underlying necrotic core defines the thin-cap fibroatheromas (vulnerable plaque) [11, 12] while more complicated plaques are represented by surface defects, and/or hematoma-hemorrhage, and/or thrombosis (AHA Type VI) [5, 8]. By histology, thin cap fibroatheromas are considered high-risk plaques with fibrous caps of thickness less than 65 μm, which are typically heavily infiltrated by macrophages and T-cells [13]. This measure of fibrous cap thickness is derived primarily from histologic sections of plaque ruptures where cap thickness at rupture sites was found to measure 23 ± 19 μm with a 95 % confidence interval of 64 μm [13]. Lesions identified as thin-cap fibroatheromas are considered precursors to rupture since they retain most features of rupture except that the fibrous cap is intact without a superimposed luminal thrombus.
The complications of hemorrhage, calcification, ulceration, and thrombosis in late stages of atherosclerosis are poorly understood. As recent as twenty-years-ago, the belief remained that acute coronary thrombosis was the sole cause of plaque rupture. Study of human coronary plaques at autopsy from sudden death victims have recently disproven this theory as our laboratory has shown three main causes of thrombosis to include plaque ruptures as the most frequent, followed by erosion, and, least frequent, eruptive nodular calcification [8].
Fatal Coronary Plaques (Fig. 1.3a–d)
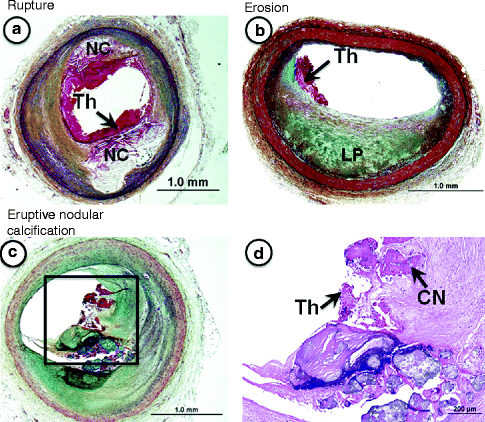
Fig. 1.3
(a–d) Lesion morphologies “fatal plaques” associated with sudden coronary death. (a) Plaque rupture shows a relatively larger necrotic core (NC) with a disrupted fibrous cap and luminal thrombus (Th), which is in communication with the NC. (b) An example of plaque erosion. The underlying eccentric plaque is consistent with pathologic intimal thickening as it contains a lipid pool (LP) in the absence of necrosis. There is a superimposed non-occlusive luminal thrombus (Th), which does not communicate with the underlying plaque. The luminal surface near the thrombus is rich is smooth muscle cells and proteoglycans. Erosions are more common in young women and smokers. (c) Shows eruptive nodular calcification, a minor but viable mechanisms of acute coronary thrombosis. (d) The relatively fibrotic plaque shows both matrix and nodular calcification (CN) where the latter are seen extending into the luminal space where there is a non-occlusive luminal thrombosis (Th). (a–c) Movat pentachrome staining; (d) Hematoxylin and eosin (H&E).
Plaque rupture is defined by fibrous cap disruption or fracture whereby the overlying thrombus is in continuity with the underlying necrotic core.
Plaque erosion is identified when serial sectioning through a thrombus fails to show communication with a necrotic core or deep intima; the endothelium is absent, and the thrombus is superimposed on a plaque substrate primarily composed of smooth muscle cells and proteoglycans.
Calcified nodule is characterized by eruptive, dense calcified bodies protruding into the luminal space, and represents the least frequent morphology associated with luminal thrombosis.
In approximately 50–60 % of sudden coronary deaths, the culprit fatal lesion exhibits an acute coronary thrombus whereas the remainder include stable coronary plaques with >75 % cross-sectional area luminal narrowing [14]. Moreover, greater than half of patients without acute coronary thrombi have healed myocardial infarcts, and, in 15–20 % of cases, there is no underlying myocardial pathology implicating a terminal arrhythmia [8].
The cause(s) of rupture are poorly understood although responsible factors involve matrix metalloproteinase expression MMPs [15], high shear regions [16], stress points, calcification and iron deposition within the fibrous cap [17]. Recent data are also beginning to unravel critical differences in gene expression between stable and unstable atherosclerotic plaques [18].
Imaging Atherosclerosis
To move the field forward in the area of lesion imaging, an understanding of the plaque microenvironment will likely provide key systemic and local signatures, which would be helpful in the recognition of the vulnerable plaque. Considering the necrotic core is a likely indicator of significant plaque progression and is a recognized feature of lesion vulnerability, it is particularly important to understand how necrotic cores form from the perspective of primary and secondary inflammation, cell death and removal of debris, and the host of other crucial factors that may be involved such as tissue disruption proteases [15] and hemorrhage [19]. Further classification of immune cells into different cellular states or subtypes may help provide further insight into their disparate functions since macrophages likely play a diverse role in disease progression beyond their primary involvement in lipid uptake.Related posts:
 Positron Emission Tomography: Imaging Inflammatory Atherosclerosis
Positron Emission Tomography: Imaging Inflammatory Atherosclerosis
 Factors Contributing to the Development of Atherosclerosis
Factors Contributing to the Development of Atherosclerosis
 Atherosclerosis: Clinical Perspectives Through Imaging Carotid Intima-Media Thickness
Atherosclerosis: Clinical Perspectives Through Imaging Carotid Intima-Media Thickness
 Intravascular Ultrasound
Intravascular Ultrasound
 Noncontrast Cardiac CT for the Detection of Calcified Atherosclerosis
Noncontrast Cardiac CT for the Detection of Calcified Atherosclerosis
 Coronary Atherosclerosis Imaging by Coronary CT Angiography
Coronary Atherosclerosis Imaging by Coronary CT Angiography

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


