Heritable Disorders of Connective Tissue
Bart L. Loeys
Michael J. Wright
Harry C. Dietz III
The diversity of clinical features in connective tissue diseases pays witness to the ubiquitous nature of connective tissue itself. Therefore, determining what is and is not a connective tissue disease can be difficult and subject to individual interpretation. We have chosen to follow the lead set in McKusick’s Heritable Disorders of Connective Tissue and Connective Tissue and Its Heritable Disorders in this matter. We recommend both these texts as excellent sources of detailed reviews regarding the normal physiology of connective tissue and the molecular basis of heritable disorders. In this chapter, we do not cover the chondrodysplasias. For further discussion of skeletal dysplasias, we refer the reader to Chapter 433 and to the excellent review by Kornak and Mundlos.
MARFAN SYNDROME AND RELATED PHENOTYPES
Marfan Syndrome
Marfan syndrome is one of the best studied and most fully understood of the connective tissue diseases. It is a dominantly inherited disorder with a birth incidence estimated at 1 in 5,000. This may be an underestimation because of the high degree of clinical variability and the lack of a fully sensitive diagnostic test.
Clinical Features
Although the genetic defect that underlies Marfan syndrome is known, the diagnosis still largely depends on the identification of specific clinical features. Those features are found mainly in the skeleton, the cardiovascular system, the eyes, the lungs, and the integument. Abnormalities of the skeleton, along with a degree of muscle hypoplasia, give rise to the distinctive appearance of individuals with Marfan syndrome (Fig. 392.1). The increased total height that may be present from birth is caused by expansion of the lower segment, measured from the symphysis pubis to the floor in an individual standing in bare feet. Consequently, the ratio of the upper segment to the lower segment is decreased as compared with controls. Usually, the ratio in individuals with Marfan syndrome is more than 2 standard deviations below the mean for age and, in general terms, is less than 1.0 at 0 to 5 years, less than 0.95 at 6 to 7 years, less than 0.90 at 8 to 9 years, and less than 0.85 in those older than 10 years. Upper extremity involvement is reflected in an increased span-to-height ratio, with greatest diagnostic significance given to values greater than 1.05. Arachnodactyly is a subjective finding that can be assessed clinically by eliciting the thumb sign (positive if the entire distal phalanx of the thumb projects beyond the ulnar border of the palm when the thumb is opposed maximally) and the wrist sign (positive if the distal phalanges of the thumb and little finger completely overlap when wrapped around the contralateral wrist). Joint laxity affects both large and small joints and may give rise to recurrent dislocation. It is particularly problematic in the feet, where pes planus and instability of the ankle can cause difficulties with ambulation. Paradoxically, limitation of movement or even joint contracture, especially of the elbow and fingers, may be present. Later in life, arthritis may develop in previously hypermobile joints. Protrusio acetabuli (deep protrusion of the femoral head into the pelvis) occurs in up to 50% of cases but rarely causes clinical manifestations.
The axial skeleton is prominently involved: scoliosis with or without kyphosis of the thoracic spine occurs in approximately 60% of cases. Spondylolisthesis may be found in the lumbosacral spine. Physiologic thoracic kyphosis can be replaced by a “straight back” or even by thoracic lordosis; in association with the pectus excavatum, this condition may give rise to significant reduction in anteroposterior diameter of the chest. Pectus excavatum and pectus carinatum arise as a result of rib overgrowth.
Abnormalities of the bones of the skull and face manifest as dolichocephaly, a long, thin face, malar hypoplasia, a high, arched palate (giving rise to dental crowding and malocclusion), and micrognathia. Other facial findings include deeply set eyes and downward slant of the palpebral fissures.
The increased mortality associated with Marfan syndrome at all ages is explained almost exclusively by cardiovascular complications. The most common cardiovascular complications are mitral valve prolapse and aortic root dilatation. Mitral valve prolapse may give rise to mitral regurgitation with the concomitant risks of congestive heart failure and dysrhythmia secondary to left atrial enlargement. Mitral valve disease is the most common indication for cardiovascular surgery and the most common source of early mortality in the pediatric population. Aortic root dilatation predisposes an affected individual not only to aortic regurgitation but to the most feared of all complications of Marfan syndrome: aortic dissection and rupture. Usually, dilatation is limited to the ascending aorta, but it can involve to the abdominal aorta and the pulmonary artery. Commonly, echocardiography is used to monitor cardiovascular involvement and for planning appropriate intervention (see the later discussion of management). Importantly, subsets of individuals show primary involvement of the descending thoracic and abdominal aorta. This may increase in incidence as people with Marfan syndrome are living longer as a result of therapeutic interventions. Computed tomographic or magnetic resonance imaging modalities should be used to monitor the descending aorta.
Subluxation or complete dislocation of the lens is the classic ocular sign in Marfan syndrome. This condition may be present at birth but can present or progress throughout childhood. Formal slit-lamp examination is essential to confirm this feature, but the presence of iridodonesis (a shimmering of the iris on accommodation) is indicative. In addition to myopia and retinal detachment secondary to increased length of the eyeball, glaucoma and cataracts occur at greater than expected frequency and earlier in life than in the general population.
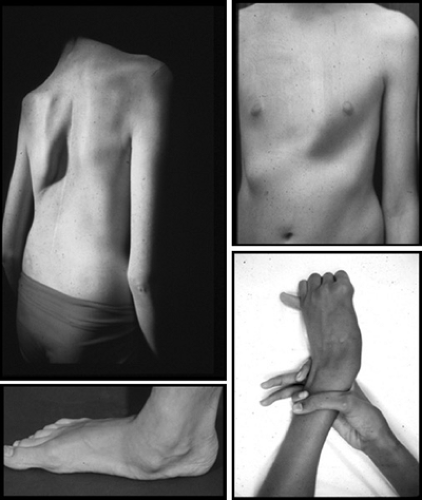 FIGURE 392.1. Major skeletal manifestations of Marfan syndrome such as scoliosis, pectus carinatum, pes planum, and positive wrist and thumb signs. |
Dural ectasia has become recognized as a feature of both diagnostic and clinical importance. In this defect, the dural sac gradually expands with erosion of sacral bone. Usually, this condition remains asymptomatic but should be considered in the differential diagnosis of back or leg pain in individuals with Marfan syndrome. Rarely, dural ectasia produces a meningocele presenting as a pelvic mass.
Despite a reported association between Marfan syndrome and attention-deficit/hyperactivity disorder, it remains unclear whether attention-deficit disorder occurs at an increased frequency in this patient population.
Striae distensae may occur, particularly after puberty, affecting the shoulders, thighs, and, often and most impressively, the lumbar region. Recurrent inguinal or incisional hernias also are common. Otherwise, the skin is involved minimally.
Often, spontaneous pneumothorax is perceived as a common complication of Marfan syndrome; in reality, it is relatively rare. Pulmonary function may be compromised by severe scoliosis and chest wall deformity.
Pathology
Aortic dilatation begins at the sinuses of Valsalva and extends into the ascending aorta and (in some cases) beyond. Commonly, dissection begins at the site of maximal dilatation. Histologically, the medial layer of the aorta is disorganized,
with fragmentation of the elastic matrix and accumulation of other matrix components. Recent evidence suggests that structural deficiency of fibrillin-1 leads to overactivation of the transforming growth factor-beta (TGF-beta) signaling pathway.
with fragmentation of the elastic matrix and accumulation of other matrix components. Recent evidence suggests that structural deficiency of fibrillin-1 leads to overactivation of the transforming growth factor-beta (TGF-beta) signaling pathway.
To date, mutations causing Marfan syndrome have been found only in the gene encoding fibrillin-1 (FBN1). Fibrillin-1 is the major component of the extracellular microfibril. Fibrillin1 molecules multimerize and aggregate with other structural proteins to form microfibrils. To date, more than 560 different mutations have been found in FBN1, precluding effective population screening.
Diagnosis
The diagnosis of Marfan syndrome is complicated by the common occurrence of a number of the features of the condition, both as isolated findings in the general community and as part of other connective tissue diseases, such as the Ehlers-Danlos syndromes and familial mitral valve prolapse syndromes (see later). In an attempt to overcome these difficulties, criteria for diagnosing Marfan syndrome were established, as reported by Beighton and associates and more recently were revised, as noted by De Paepe and colleagues. The discovery of the molecular basis of Marfan syndrome, as reported by Dietz and colleagues, has not allowed for the creation of a definitive and universally applicable diagnostic test, because of the large number of separate causative mutations and the finding that related but distinct conditions can be caused by mutations in the same gene. Linkage analysis can identify at-risk individuals and pregnancies in families of sufficient size even if a mutation is not known, but discovery of a pathogenic mutation renders this possible regardless of family size.
Therefore, diagnosis in most cases is based on the clinical assessment and criteria shown in Table 392.1. In addition to careful history and clinical examination, the only specialized investigations required are slit-lamp examination, echocardiography, and plasma amino acid analysis (to exclude the diagnosis of homocystinuria). The diagnosis of Marfan syndrome in an individual with no family history requires identification of major criteria in two organ systems, with involvement of a third. If a major criterion is established by virtue of family history, major criteria must be met in one organ system and another must be involved.
TABLE 392.1. DIAGNOSTIC CRITERIA FOR MARFAN SYNDROME* | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Management
Management of aortic root dilatation or dissection, aortic regurgitation, and mitral valve abnormalities is important because appropriate treatment can have significant effects on morbidity and mortality. Echocardiographic measurement of aortic root diameter at the sinuses of Valsalva and assessment of aortic and mitral valve function should be carried out at least yearly. More frequent evaluations may be necessary with severe regurgitation or aortic dilatation, or with rapid rate of growth of the aorta. Charts of aortic root diameter plotted against body surface area are available for normalization, as reported by Roman and associates. Surgical intervention should proceed at any age when the maximal aortic root diameter reaches 5.0 cm. This guideline is based on the observation of aortic dissection in older children or adults that cross this threshold. Given the extreme rarity of aortic complications in young children, it has been difficult to establish rules with similar predictive value and conviction. Current consensus suggests that surgery should be considered in young children who develop severe aortic regurgitation or aortic root growth that exceeds 1 cm/year. Indications based on aortic size have been impossible to validate, and the recent trend has been to tolerate aortic dimensions up to 5.0 cm irrespective of age. It is anticipated that these guidelines will be refined with further experience.
Various surgical techniques have been developed, but recently the results of valve-sparing procedures have been quite encouraging. This procedure avoids lifelong anticoagulation and is particularly appealing for young woman who anticipate pregnancy. Use of a homograft remains an alternative for very young children who require surgery. Given the primary involvement of the pulmonary artery, the Ross procedure, which involves replacing the aortic root with a pulmonary autograft, is contraindicated in Marfan syndrome and in other connective tissue disorders. Severe left ventricle enlargement or dysfunction and valve insufficiency are indications for surgical repair or replacement of the aortic or mitral valves.
Beta blockade has been shown to slow the rate of aortic root dilatation and to reduce the incidence of aortic dissection in Marfan syndrome. Our policy has been to treat all individuals, including children with a confirmed diagnosis of Marfan syndrome, with atenolol in sufficient doses to maintain the heart rate after submaximal exercise at less than 110 beats/minute. This treatment is generally well tolerated in children and adults with Marfan syndrome. Treatment with verapamil should be considered in individuals who do not tolerate beta blockade.
Individuals with Marfan syndrome should remain active with aerobic activities performed in moderation. Contact sports, competitive sports, exercise to the point of exhaustion, and isometric exercises (e.g., weight lifting) should be avoided because of the increased risk of aortic dilatation, dissection, or rupture. Antibiotic prophylaxis should be liberally prescribed, because the inherent abnormality of the valve tissue (e.g., myxomatous mitral valve changes) can predispose patients to bacterial endocarditis.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


