Hereditary and Acquired Types of Myopathy
Darryl C. De Vivo
Salvatore DiMauro
Molecular genetics has revolutionized our current understanding of neuromuscular diseases. The inherited conditions embrace all patterns of heredity, both mendelian and nonmendelian. The term muscular dystrophy is used to describe a group of conditions that are inherited, progressive, and characterized by myodegeneration. Other conditions are designated as types of myopathy, such as congenital myopathy (e.g., central core disease) or metabolic myopathy. In general, forms of metabolic myopathy are classified according to the involved pathway (e.g., types of glycogenoses, forms of mitochondrial myopathy), and often the specific biochemical defect is known. Whenever possible, these diseases are classified according to the molecular, genetic, or biochemical defect because the phenotypic expression of one molecular defect may be fairly heterogeneous. Thus, Duchenne-type muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are allelic diseases, whereas McLeod syndrome and the dystrophy associated with glycerol kinase deficiency are due to contiguous genes involvement. We know now that the heterogeneous phenotypes associated with a specific genetic defect may result from the variable involvement of contiguous genes, depending on the size of the deleted genetic segment.
The acquired muscle disorders are classified according to etiologic factors whenever possible or by the characteristics of the tissue reaction. Hence, we use the terms inflammatory, toxic, nutritional, and endocrinologic to describe these diverse conditions.
HEREDITARY TYPES OF DYSTROPHY
X-Linked Diseases
Duchenne Muscular Dystrophy
DMD is the most severe form of progressive primary muscular degeneration. It affects approximately one in 3,000 live-born male infants, with one-third of all cases representing new mutations. DMD is associated with mutations in a gene located in band 1 of region 2 of the short (p) arm of the X chromosome (Xp21), which encodes for a large protein called dystrophin. DMD is manifest at birth, becomes clinically evident when children are between 3 and 5 years of age, and progresses inexorably over the course of the next 2 decades before culminating in death. Most patients become wheelchair-dependent when they are between 10 and 12 years of age.
Pathogenesis
DMD is associated with mutations in a gene located in band 1 of region 2 of the short (p) arm of the X chromosome (Xp21), which encodes for a large protein called dystrophin. The genetic cause of DMD is known, but the pathophysiology remains obscure. The dystrophin gene—the largest identified thus far—encompasses 80 exons, with multiple spliced isoforms and many tissue-specific promoters. The typical DMD phenotype is due to lack of the full-length muscle dystrophin transcript that encodes a dystrophin isoform of 427 kd. The most common gene defects associated with DMD are deletions involving one or more exons. The severity of the clinical phenotype depends largely on how a deletion affects the reading frame. If the “residual” gene still can be spliced together into an abnormal but still “readable” mRNA, then an abnormal dystrophin is made and the clinical presentation is milder, usually BMD (see section, Becker Muscular Dystrophy). If, however, the deletion causes a frame shift and generates an unreadable mRNA, then no dystrophin is made and clinical presentation is severe DMD.
Dystrophin, with a molecular weight of 440 kd, is a central component of the membrane cytoskeleton, in close association with other proteins, including sarcoglycans, dystroglycans, and laminin (Fig. 409.1). This complex confers structural integrity to the muscle plasma membrane, which is subject to the mechanical stress of contraction. Thus, a simple—perhaps simplistic—pathogenic hypothesis is that “tears” in the plasma membrane lead to cell death. However, this mechanism does not explain the presymptomatic period or the progressive nature of the disease. Other hypotheses consider the partial compensatory roles of other cytoskeletal proteins (such as utrophin), the limited regenerating potential of continually damaged myofibers, and the potential contribution of inflammation and fibrosis. The histopathologic abnormalities seen in the various types of DMD are distinctive but are not specific for any entity (Box 409.1).
Clinical Manifestations and Complications
Infant macroglossia has been noted occasionally, and motor milestones may be delayed. One-third of patients with DMD are late walkers (i.e., they do not walk independently until they are between 15 and 18 months of age). Parents retrospectively report developmental clumsiness and motor sluggishness in running, climbing stairs, rising from the ground after falling, and pedaling a tricycle. Abnormalities of gait and posture appear in middle childhood, with the emergence of increasing lumbar lordosis, pelvic waddling, frequent falling, and Gowers sign. Although Gowers sign is distinctive in DMD, it may be seen in patients with any condition that causes pelvic girdle weakness. Enlargement of the musculature becomes evident, with characteristic involvement of calf, gluteal, lateral vastus, deltoid, and infraspinatus groups. Weakness is more evident in the proximal muscles, and tendon reflexes are diminished at the knees, biceps, and triceps. Only in the preterminal phase are the distal tendon reflexes affected noticeably. Before ambulation is lost, contractures of the iliotibial bands, hip flexors, and heel cords develop. After ambulation is lost, the muscles decrease in size, contractures progress with loss of joint mobility, and kyphoscoliosis develops with further compromise of respiratory function.
Complications may result from the following:
Cardiac involvement is evident in all patients with DMD but rarely is the cause of death. Similarly, cardiac abnormalities may be noted in female carriers of DMD, even when the serum creatine kinase (CK) values are normal. Degenerating muscle fibers and small foci of fibrosis are scattered throughout the myocardium and conduction systems. The posterobasal region and adjacent lateral wall of the left ventricle are involved commonly and prominently. The changes in electrocardiogram (ECG) are distinctive: tall right precordial R waves and deep Q waves in the left precordial and limb leads.
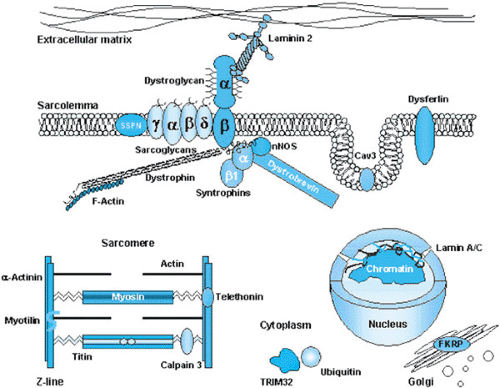
FIGURE 409.1. Schematic representation of the proteins involved in the muscular dystrophies. (Reproduced with permission from Barresi R, Campbell KP. Limb-girdle muscular dystrophies. In: Rosenberg RN, Prusiner SB, DiMauro S, et al, eds. The molecular and genetic basis of neurologic and psychiatric disease, 3rd ed. Boston: Butterworth-Heinemann, 2003:479.)
Nervous system involvement has been recognized since the earliest descriptions of DMD. It is a nonprogressive process and may be associated with the “atrophy” of the brain seen on computed tomography (CT). Some patients also have macrocephaly. The mean IQ is approximately 80, and individual IQ values correspond to a gaussian, bell-shaped distribution curve.
BOX 409.1 Histopathology of Duchenne Muscular Dystrophy
Necrosis and regeneration dominate the histopathologic findings in biopsy samples from young patients with DMD, and end-stage biopsy samples obtained from patients with preterminal disease reveal the replacement of muscle cells with fat cells and fibrous tissue. Groups of necrotic degenerating fibers are the most prominent early features in DMD. These clusters of necrotic cells are invaded by phagocytes, and the infiltration is followed by active regeneration. A variation in fiber size becomes evident, with small atrophic fibers located adjacent to hypertrophied elements. Some of the large fibers are hypercontracted or have undergone hyalinization. Endomysial and perimysial connective tissue gradually increases with progression of disease. Plasma membrane defects have been seen by electron microscopy in non-necrotic muscle fibers and are thought to represent an early or possibly basic change in DMD. The basal lamina overlying the plasma membrane defect is normal.
Diagnosis
The differential diagnosis of DMD includes BMD, autosomal recessive forms of congenital muscular dystrophy or limb-girdle dystrophy (see later discussion), muscle carnitine deficiency, the childhood form of acid maltase deficiency, and juvenile spinal muscular atrophy (Kugelberg-Welander syndrome). Female patients with clinical features of DMD must undergo genetic and cardiologic examination. Such patients may represent sporadic cases of autosomal recessive limb-girdle dystrophy, manifesting carriers of DMD, or genuine examples of DMD resulting from selective chromosomal aberrations. These aberrations include Turner syndrome (XO karyotype), mosaic states (X/XX or X/XX/XXX), a structurally abnormal X chromosome, an X autosomal translocation, and, perhaps more important, a skewed inactivation of the X chromosome favoring the one carrying the mutant gene.
The diagnosis of DMD can be made reliably in virtually every case using currently available information. The clinical presentation and course are constant in most instances. The CK value is very high in the preclinical phase and falls gradually as the muscle mass disappears in later years. The electromyogram (EMG) and ECG findings are distinctive. The EMG is distinctively myopathic, with decreases seen in the amplitude and duration of the compound action potential and enrichment of the interference pattern. A large number of the motor units are polyphasic, and occasional sparse fibrillation potentials are observed consistently. Sensory and motor conduction velocities are normal, and morphologic findings on biopsy of the skeletal muscle are characteristic. A combination of dystrophin immunoreactivity and Western blot analysis of muscle obtained at biopsy or analysis of blood DNA finalizes the diagnosis. This constellation of clinical and laboratory findings permits the exclusion in almost every case of those diseases that masquerade clinically as DMD. The emerging molecular and biochemical advances have added measurably to our understanding of the phenotypic expressions of diseases associated with a genetic defect at the Xp2.1 locus, collectively called dystrophinopathies.
Therapy
Fifteen years after the discovery of the gene defect in DMD, therapy remains inadequate. Multiple pharmacologic approaches, aimed at blocking the pathophysiologic pathway in humans or in animal models (especially the mdx mouse), have been largely unsuccessful. Attempts to replace the defective protein by direct transfer of normal stem cells into affected muscles—myoblast transfer—has had limited success in mdx mice. The systemic delivery of muscle stem cells is being attempted in animal models. Direct gene replacement (gene therapy) is daunting because of the enormous size of the gene, the need for appropriate vectors, and the risks of immunologic reaction occurring. However, the use of truncated genes (minigenes) carried by adeno-associated virus (AAV) has shown some success in animals. Thus, for now, traditional methods of care remain important. Family counseling, physical therapy, proper use of orthotic devices, selective surgical interventions to treat joint contractures and spinal deformities, and dietary management are important ways to improve the quality and length of life for patients with DMD.
Clinical trials of promising therapeutic regimens have been conducted by the Collaborative Investigation of Duchenne Dystrophy (CIDD) group sponsored by the Muscular Dystrophy Association. The CIDD has documented the clinical course of DMD carefully and has developed rigorous protocols to assess the efficacy of various drugs and hormones used in its treatment. Treatment with prednisone, 0.75 mg/Kg/day, has been shown in double-blind, placebo-controlled trials to slow the decline of muscle strength and to increase muscle mass.
Becker Muscular Dystrophy
Two other disorders share a genetic defect at or near the Xp2.1 locus: BMD and the McLeod syndrome. BMD represents a more benign version of DMD, most commonly due to a deletion in the dystrophin gene that does not alter the reading frame of the transcript (as described earlier, in the section Duchenne Muscular Dystrophy). Current information indicates that BMD and DMD are allelic gene abnormalities. BMD is similar to DMD, but the age of onset is later and the progression is slower (Table 409.1).
Pseudohypertrophy is striking, and frequently pes cavus deformities are present in patients with BMD. Unlike in DMD, cardiac or nervous system involvement is an unusual finding, but “pure” dilated cardiomyopathy can occur in patients with BMD in the absence of muscle weakness. Other unusual clinical phenotypes include recurrent myoglobinuria or idiopathic hyper-CKemia, and weakness confined to the quadriceps (quadriceps myopathy). Patients with BMD may have children, although infertility rates are higher in this population. All female progeny are obligate carriers, and all sons are unaffected.
TABLE 409.1. DISEASE-RELATED EVENTS IN BMD AND DMD | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
The EMG in patients with BMD is distinctly myopathic, but neurogenic features also may be present. The results of muscle biopsy are compatible with a dystrophic process, but neurogenic atrophy may be seen in one-half of the cases. The neurogenic features are thought to result possibly from muscle fiber splitting. Dystrophic immunoreactivity is present in muscle obtained on biopsy, but the subsarcolemmal staining pattern demonstrates discontinuities. Western blot analyses often show dystrophin of increased or decreased size. The serum CK values are elevated, but to a less striking degree than those in patients with DMD.
Differentiating between BMD and limb-girdle muscular dystrophy may be difficult in sporadic cases. Often, calf hypertrophy is more marked and persistent in patients with BMD. Loss of ambulation is the single best discriminator between DMD and BMD (see Table 409.1). Contractures, a rigid spine, and the absence of pseudohypertrophy help to distinguish Emery-Dreifuss muscular dystrophy from BMD. The treatment approaches and genetic counseling are similar to those for DMD.
McLeod Syndrome
The McLeod syndrome has been viewed by some physicians as the minimal expression of the Xp2.1 gene abnormality. The gene encoding the Kx antigens in the Kell red blood cell antigen system is contiguous to the DMD locus. McLeod syndrome is manifested by absence of the Kx antigens, hemolytic anemia, acanthocytosis, elevated serum CK activity, and a benign nonprogressive myopathy. The skeletal muscle biopsy reveals muscle fiber necrosis and regeneration. One classic patient had the genetic misfortune of having chronic granulomatous disease, McLeod red cell phenotype, DMD, retinitis pigmentosa, idiopathic intestinal pseudo-obstruction, and mental retardation. The McLeod syndrome may be confused with polymyositis.
Emery-Dreifuss Syndrome
Emery-Dreifuss syndrome also is inherited as an X-linked recessive condition, but autosomal dominant or recessive inheritance also is seen in a minority of patients. This disease begins in early childhood and progresses very slowly. Weakness is evident first in the legs, with later involvement of the pectoral girdle. Contractures of the elbows and heel cords are prominent, and early features and rigidity of the spine also may be present. Distal muscles are spared, and no pseudohypertrophy ensues. Atrial conduction abnormalities may occur and, ultimately, the cardiac involvement may result in sudden death. Serum CK values are elevated moderately. Scoliosis and hyperextension of the cervical vertebrae are conspicuous features in the rigid spine syndrome, a form of congenital muscular dystrophy. The gene responsible for Emery-Dreifuss syndrome at Xq28 contains only six exons and five introns. It encodes a 254-amino acid serine-rich protein called emerin, which is
localized to the inner nuclear membrane. Patients with autosomal dominant or autosomal recessive forms of Emery-Dreifuss syndrome harbor mutations in the lamin A/C gene (LMNA), which encodes lamins A and C: like emerin, both lamin A and lamin C are components of the nuclear lamina.
localized to the inner nuclear membrane. Patients with autosomal dominant or autosomal recessive forms of Emery-Dreifuss syndrome harbor mutations in the lamin A/C gene (LMNA), which encodes lamins A and C: like emerin, both lamin A and lamin C are components of the nuclear lamina.
Autosomal Types of Dystrophy
The generalized types of dystrophy that are inherited in an autosomal manner include the various forms of myotonia, periodic paralyses, limb-girdle dystrophies, and congenital dystrophies.
Myotonia
Myotonia is divided into two categories: progressive and nonprogressive. Myotonic dystrophy is the only progressive disorder. The nonprogressive disorders include myotonia congenita, paramyotonia, and numerous atypical myotonic disorders.
Myotonic dystrophy type 1 follows a pattern of autosomal dominant inheritance and has been subdivided into a “classical form” (Steinert disease) or dystrophia myotonica type 1 (DM-1) and two proximal myotonic dystrophy syndromes (PROMM), DM-2/PROMM and DM3/PROMM. In DM-1, gene mapping studies have localized the defect to the long arm of chromosome 19 and characterized it as an unstable CTG repeat expansion in the noncoding region of a serine-threonine protein kinase (DMPK) gene. This trinucleotide amplification correlates with the clinical phenomenon of anticipation. The clinical manifestations of myotonic dystrophy are protean, and this disorder is viewed more properly as a systemic disorder with muscle involvement. Virtually all systems are involved. Severe brain involvement may occur in the congenital form, and adults have mental deterioration. Smooth, striated, and cardiac muscle tissues are affected. Myotonia involves striated muscle and anal sphincter; smooth-muscle involvement has been seen in the gastrointestinal tract, gallbladder, uterus, ureter, and ciliary body of the eye. Endocrine disturbances include testicular tubular atrophy, pituitary dysfunction, and diabetes mellitus associated with peripheral insulin resistance. Baldness and cataracts are common manifestations, as is hypogammaglobulinemia. Cataracts are represented as multicolored subcapsular opacities visualized best by slit-lamp examination.
In virtually all affected infants with congenital myotonic dystrophy, the mother is the affected parent, despite the fact that the gene is transmitted as an autosomal dominant trait. Numerous complications of pregnancy have been recognized, including an increased rate of spontaneous abortion, reduced fetal movements, polyhydramnios, uterine inertia during labor, and postpartum hemorrhage (often associated with retained placental fragments). The neonatal mortality rate is increased, and affected infants display numerous abnormal features. Neonatal respiratory distress, paralyzed diaphragm, hypotonia, bilateral facial weakness, talipes, and delayed motor and mental retardation are common clinical features. Myotonia is conspicuously absent and is not clinically evident until later in the patient’s first decade of life. Congenital hip dislocation and hernias are the result of muscular laxity. Mild mental retardation is evident in most, if not all, affected children, and hydrocephalus has been reported in some instances. The correlation between maternal myotonic dystrophy and the congenital form of the disease is overwhelming but unexplained.
The muscle histopathology of myotonic dystrophy is distinctive. Type I fiber atrophy and centrally placed nuclei are characteristic, even in the congenital form. These and other findings suggest an arrest in muscle development: hypoplasia rather than active degeneration. Other common features include ringed fibers, sarcoplasmic masses, and nuclear chains.
Myotonic dystrophy type 2 (DM-2/PROMM) typically starts in adults and is characterized by myotonic stiffness or weakness of proximal leg muscles or finger flexors. Myalgia is a common finding; it is unrelated to exercise or to the severity of myotonia and typically fluctuates in intensity. Unlike patients with DM-1, patients with DM-2/PROMM do not show facial muscle weakness and wasting or distal limb muscle wasting. Myotonia is elicited best by percussion of forearm or thenar muscles. Cataracts are common findings. The gene defect on chromosome 3q21 is an unstable quadruplet (CCTG) repeat expansion in the gene for the zinc finger 9 protein (ZNF9). Some families with autosomal dominant inheritance of very similar symptoms and signs do not show linkage to the DM-1 or the DM-2 loci, and their gene defect is unknown: this form of myotonic dystrophy is called DM-3/PROMM.
Treatment of the various types of myotonia is directed toward their symptoms and toward the debilitating features of myotonic dystrophy specifically. The myotonia may be relieved with quinine sulfate, procainamide, or phenytoin. Patients with myotonic dystrophy need general medical supervision and support. Cardiac conduction disturbances and sensitivity to various anesthetics may be life-threatening. Medical and rehabilitative intervention, insertion of cardiac pacemakers, treatment of respiratory problems, and the use of various orthotic devices are helpful.
Periodic Paralyses, Cardiac Arrhythmias, Congenital Myotonia, and Malignant Hyperthermia
These clinically diverse conditions all are caused by mutations in genes encoding subunits of ion channels (sodium, calcium, or potassium) and, therefore, often are labeled collectively as channelopathies.
Periodic Paralyses
Often, the periodic paralyses are associated with high or low serum potassium values. The primary periodic paralyses are inherited as an autosomal dominant trait, whereas the secondary disorders result from acquired conditions that perturb body water and electrolyte status or thyroid function. The genetically determined conditions are associated with modest disturbances of potassium homeostasis, in contrast to the nongenetic conditions. Thyrotoxic periodic paralysis resembles hypokalemic periodic paralysis. It occurs remarkably more commonly in men (6:1) and Asians (3:1). The condition is sporadic and resolves with effective treatment of the hyperthyroidism.
Traditionally, the primary periodic paralyses have been divided into hypokalemic, hyperkalemic, and normokalemic. A fourth (rarer) form of periodic paralysis is associated with a bidirectional ventricular tachycardic dysrhythmia.
Hypokalemic periodic paralysis presents in middle to late childhood. Attacks are accompanied by a modest fall in the level of serum potassium and in the urinary retention of sodium, potassium, chloride, and water. These electrolyte changes can produce a characteristic change on the ECG. The attacks, which affect limb and trunk muscles but typically spare bulbar, respiratory, and cardiac muscles, may be provoked by the ingestion of carbohydrate or sodium or by excitement. Initially, the attacks occur infrequently, but daily attacks may occur during early adulthood. The episodes decrease in frequency in late adulthood, but older patients may have a fixed limb weakness. Glucose-insulin infusion may provoke an attack, and oral potassium salts attenuate the episode. These findings are diagnostic of the condition. Two common mutations have been identified in the L-type calcium channel subunit gene (CACNA1S) located on chromosome 1q31.
Hyperkalemic periodic paralysis also is inherited as an autosomal dominant trait, with initial presentation occurring during the patient’s first or second decade of life. The serum
potassium value rises modestly during the attack, accompanied by a kaliuresis. Between paralytic episodes, some patients have myotonia involving facial, lingual, thenar, or finger extensor muscles. Primary hyperkalemic periodic paralysis can be provoked by the oral administration of potassium chloride. Mutations in the sodium channel gene (SCN4A) have been identified in patients with hyperkalemic periodic paralysis.
potassium value rises modestly during the attack, accompanied by a kaliuresis. Between paralytic episodes, some patients have myotonia involving facial, lingual, thenar, or finger extensor muscles. Primary hyperkalemic periodic paralysis can be provoked by the oral administration of potassium chloride. Mutations in the sodium channel gene (SCN4A) have been identified in patients with hyperkalemic periodic paralysis.
Long QT syndrome and idiopathic ventricular fibrillation is a channelopathy affecting cardiac muscle and characterized by delayed repolarization, showing in surface ECGs as a prolonged QT interval. This important and not uncommon disorder (1 in 7,000 persons) causes ventricular arrhythmias with syncope or sudden death occurring in young asymptomatic individuals, often during exercise. Long QT syndrome is transmitted as an autosomal dominant (rarely recessive) trait and is caused by mutations in the various genes encoding subunits of the potassium channel. Muscle biopsy often reveals few or an absence of abnormalities in patients, with the periodic paralyses manifesting during the early symptomatic years, even during a paralytic episode. Later, a distinctive vacuolar myopathy develops. Often, these biopsy results correlate with fixed limb weakness.
Lack of electrical excitability of muscle fiber surface membranes is common to the various forms of primary periodic paralysis, and an abnormality of the sodium channel that characterizes the electrophysiologic study results in patients with paramyotonia congenita and those with hyperkalemic periodic paralysis (in agreement with molecular genetic data). The resting membrane potential, studied in vitro, is reduced in patients with primary hypokalemic periodic paralysis and in those with the acquired form associated with thyrotoxicosis. Both insulin and thyroid hormone are known to increase the activity of the sodium-potassium pump, but this information does not facilitate an understanding of the clinical symptomatology associated with modest decreases in serum potassium values.
Treatment begins with accurately establishing the diagnosis of the syndrome. Aggravating environmental factors must be avoided to minimize the frequency and intensity of the attacks. Acetazolamide is the preferred drug for both the hypokalemic and hyperkalemic forms of primary periodic paralysis; it may be useful also in the paralysis associated with cardiac dysrhythmias. However, treatment of the paralysis may worsen the cardiac disturbance. Imipramine has been useful in one patient in controlling the dysrhythmia. Acetazolamide is ineffective in treating the paralysis associated with hyperthyroidism. Propranolol is helpful, but control of the thyroid disease is more important.
Congenital Myotonia
The nonprogressive types of myotonia as a group are rare, and the symptomatology is limited to skeletal muscle, unlike that of myotonic muscular dystrophy. Myotonia congenita is an autosomal dominant trait that appears in infants. Muscular hypertrophy is evident. Becker later distinguished the autosomal dominant (Thomsen) form of nonprogressive myotonia from the recessive form, in which generalized myotonia first appears in early to middle childhood. Muscular hypertrophy is even more striking in the recessive form of the disease. Subtle weakness also may be present. Both the dominant and the recessive forms of myotonia congenita result from mutations in the skeletal muscle chloride channel CLCN1 gene. Disturbances of chloride conductance, a crucial factor in the repolarization and electrical stability of the sarcolemma, explain the myotonic runs observed on EMG.
Potassium-aggravated myotonia refers to a childhood-onset syndrome resembling myotonia congenita, without associated weakness or periodic paralysis. The myotonia is alleviated by repeated contractions (“warm-up” phenomenon) and worsened by potassium intake. Some patients have hypertrophy of neck and shoulder muscles. In children, potassium-rich meals may cause hypoventilation due to stiffness of thoracic muscles. This condition has been associated with mutations in the sodium channel gene (SCN4A).
Paramyotonia congenita has two clinical signatures: (a) it is paradoxically worsened by repetitive exercise (the reverse of the “warm-up” phenomenon) and (b) it is triggered by exposure to cold. Onset is during infancy, and inheritance is autosomal dominant. Episodes of paralysis may be induced by intake of potassium, similar to hyperkalemic periodic paralysis. As in hyperkalemic periodic paralysis, mutations affect the sodium channel gene (SCN4A).
Malignant Hyperthermia
Malignant hyperthermia has occurred in association with several of the forms of myopathy discussed, especially central core disease (discussed more fully in the section, Congenital Myopathies). However, most patients with malignant hyperthermia have no obvious clinical symptoms of muscle disease, and the EMG and muscle histology results are normal or nonspecifically abnormal. These latter patients do have an underlying myopathy that may be manifested by increased serum CK values or in vitro studies that document sensitivity to caffeine or anesthetic agents. Malignant hyperthermia is inherited in most patients in an autosomal dominant pattern. However, this condition may not be recognized until the patient is exposed to such anesthetic agents as halothane or succinylcholine, and such exposure may be catastrophic. A family history of an untoward anesthetic experience, recurrent myoglobinuria, or sudden death should suggest the possible presence of this muscle condition.
The molecular basis of malignant hyperthermia has been clarified, at least partially. More than 50% of autosomal dominant families harbor mutations in the gene (RYR1) that encodes the sarcoplasmic reticulum calcium release channel or ryanodine receptor. Mutations in RYR1 may cause malignant hyperthermia or central core disease, which are allelic diseases for this locus and can occur together in the same family. A smaller number of families with malignant hyperthermia harbor mutations in genes for calcium channel subunits: CACNA1S, encoding the alpha-1-subunit of the voltage-dependent L-type calcium channel (malignant hyperthermia linked to this locus is allelic to hypokalemic periodic paralysis), or CACNL2A, encoding the alpha-2-subunit of the same channel.
The symptom complex is characterized by rapidly rising body temperature, tachycardia, tachypnea, cyanosis, and respiratory and metabolic acidosis. Usually, affected patients experience rigidity and myoglobinuria. Unexplained fevers, muscle cramps, and increased serum CK values may represent clinical clues to the presence of malignant hyperthermia. The clinical syndrome can be aborted by the intravenous administration of dantrolene.
Congenital Muscular Dystrophies
Many primary muscle diseases may present in early infancy, but only a few are known as congenital types of muscular dystrophy. This distinction, therefore, is arbitrary and, to some degree, confusing. The conditions discussed here are two syndromes that cause dystrophic changes in skeletal muscle, as noted by biopsy. These syndromes are distinguished by the presence or absence of nervous system pathology.
Congenital muscular dystrophy without nervous system involvement (also called classical, occidental, or pure CMD) appears to be inherited as an autosomal recessive trait, although many cases are sporadic. Affected infants are weak at birth, and often hypotonia is present, although not invariably. Similarly, joint contractures frequently accompany the weakness (arthrogryposis multiplex congenita). Respiratory difficulties may be present and largely determine the outcome. The “dystrophic” process itself is rather stationary, in part controverting the use of the term. Affected infants do reasonably well if they survive
past infancy. Weakness does not progress in any measurable way, but orthopedic complications may supervene and require therapeutic intervention.
past infancy. Weakness does not progress in any measurable way, but orthopedic complications may supervene and require therapeutic intervention.
The laboratory abnormalities are variable. CK values are elevated, but tend to decrease with time. The EMG is distinctly myopathic, and fibrillations are scarce. The muscle tissue is abnormal, with fiber diameter variability and an abundance of endomysial and perimysial connective tissue. Necrosis of single muscle fibers is a fairly unusual occurrence.
The clinical course of affected patients is influenced largely by the respiratory complications that develop. The muscle disease appears to be primarily nonprogressive in most patients, and many of these children are very bright. Aggressive support and orthopedic corrective surgery are warranted in any children who have survived past infancy.
Approximately one-half of patients with typical congenital muscular dystrophy show deficiency of laminin-2, also known as merosin, a protein that links the dystroglycan complex of the sarcolemma with the extracellular matrix (see Fig. 409.1). The dystroglycans, together with dystrophin and with the sarcoglycans, are essential in maintaining the integrity of muscle membranes during the harsh mechanical stress imposed by muscle contraction. For additional information about merosin in CMD, see Box 409.2.
Congenital muscular dystrophy with nervous system pathology has a less favorable prognosis. In Japan, the birth incidence of Fukuyama syndrome is approximately one-half that of DMD. Mothers of affected infants report decreased fetal movements, and the newborn is weak and hypotonic. Sucking efforts and crying are weak, and facial expression is decreased. The calves may be enlarged, and contractures of the knee joints and elbows are common findings; tendon reflexes are decreased or absent. Generalized convulsions may occur in one-half of affected patients, and motor retardation is evident during later development. Eventually, many children sit or crawl, but few are able to walk. Atrophy of limb muscles is increasingly evident, and most affected children die within their first decade.
BOX 409.2 Merosin and Congenital Myotonic Dystrophy
The gene for the protein merosin is located on chromosome 6q2, and several mutations have been identified. Several children with merosin-deficient CMD have white matter abnormalities on cerebral magnetic resonance imaging (MRI), typically, white matter hypodensity on T2-weighted images. Otherwise, the brain usually is structurally normal. Immunohistochemistry of muscle shows more or less severe deficiency of laminin alpha-2 (for an accurate diagnosis, it is important to use antibodies to different portions of the laminin alpha-2 chain). Identification of mutations in the LAMA2 gene confirms the diagnosis.
Merosin-positive patients with CMD are a heterogeneous group, but certain clinical features allow division into distinctive subgroups that are associated increasingly with specific gene defects. Thus, linkage analysis in some families with CMD and rigid spine syndrome (caused by axial muscle contractures) has identified a locus on chromosome 1 (RSMD1), and the sequence of candidate genes has revealed pathogenic mutations in a gene encoding a selenoprotein (SEPN1). Another form of merosin-positive CMD, Ullrich muscular dystrophy, is characterized by distal hyperextensibility contrasting with proximal joint contractures: the gene responsible for Ullrich disease (COL6A3) encodes subunits of type VI collagen.
The association of Fukuyama muscular dystrophy and xeroderma pigmentosum led to the identification of the locus for Fukuyama muscular dystrophy on chromosome 9q31–33. The gene has been identified, and at least two independent point mutations have been identified in affected individuals. The gene encodes a novel protein called fukutin, which is expressed in various tissues in normal individuals but not in patients in whom the mutated gene contains a retrotransposal insertion of sequences repeated in tandem.
The serum CK values are elevated ten- to 50-fold in most instances, and the EMG is distinctly myopathic. The skeletal muscle examined at biopsy is abnormal, with variation of fiber-type diameter, necrosis of fibers, scattered foci of inflammation, and increased endomysial and perimysial connective tissue. The neuropathology is equally remarkable and descriptive of altered neuronal migration and cortical sulcation. The cortex is thick and smooth, particularly in the temporal and occipital regions. These findings are indicative of lissencephaly or polymicrogyria. Reduction in central white matter and associated cerebral ventricular enlargement are evident.
Another condition combining congenital muscular dystrophy and congenital brain abnormalities is the Muscle-Eye-Brain disease (MEB), first described in Finland, where the disease is encountered most frequently. Patients have congenital hypotonia, mental retardation, and visual failure, including severe progressive myopia, retinal degeneration, and optic atrophy. Seizures are common manifestations. Dystrophic changes occur in muscle, with normal immunohistochemistry for dystrophin but often secondary partial defect of laminin alpha-2. Brain abnormalities include cobblestone lissencephaly, frontal pachygyria, and occipital micropolygyria. White matter is affected minimally. After the locus for MEB was assigned to chromosome 1p32–34 by linkage analysis and homozygosity mapping, mutations were identified in a gene (POMGnT1) that encodes a glycosyltransferase. A third condition, clinically similar to MEB, is Walker-Warburg Syndrome (WWS; MIM 236670). WWS is inherited as an autosomal recessive trait and characterized clinically by ocular abnormalities, muscular dystrophy, and severe neuronal migration defects. The condition results from mutations in the O-mannosyltransferase gene POMT1. These recent studies suggest that glycosylation defects are important causes of congenital muscular dystrophies and that overexpression of the gene LARGE, which encodes a glycosyltransferase, can circumvent the defective glycosylation of alpha-dystroglycan in cells from patients. This observation opens new vistas on therapeutic approaches.
Localized Types of Dystrophy
Many forms of muscular dystrophy are characterized by localized muscular involvement. These syndromes may involve the limb-girdle musculature preferentially, facioscapulohumeral (FSH) groups, scapular and peroneal groups, distal limb musculature, extraocular muscles, or oropharyngeal muscles. Most of these categories are fairly heterogeneous, and phenotypic features are varied.
Limb-Girdle Syndromes
In 1954, Walton and Nattrass introduced the term limb-girdle muscular dystrophy to describe, in a number of patients, a disorder that did not fulfill the clinical criteria for DMD or FSH dystrophy. Affected patients were of both genders, often were in middle to late childhood, and had serum enzyme, EMG, and skeletal muscle biopsy abnormalities of the type commonly encountered in patients with a muscular dystrophy.
This large and ill-defined group of patients has been subdivided into 15 entities associated with specific gene defects. The conventional nomenclature assigns the number 1 (LGMD1) to the forms transmitted by autosomal dominant inheritance and the number 2 (LGMD2) to the autosomal recessive forms. The capital letters that follow (LGMD1A-E and LMGD2A-J) identify specific gene defects (Table 409.2). Figure 409.1 shows the proteins involved in these and other muscular dystrophies. Although LGMDs traditionally are considered diseases of adults, many of them can start in childhood and are outlined in Table 409.2.
This large and ill-defined group of patients has been subdivided into 15 entities associated with specific gene defects. The conventional nomenclature assigns the number 1 (LGMD1) to the forms transmitted by autosomal dominant inheritance and the number 2 (LGMD2) to the autosomal recessive forms. The capital letters that follow (LGMD1A-E and LMGD2A-J) identify specific gene defects (Table 409.2). Figure 409.1 shows the proteins involved in these and other muscular dystrophies. Although LGMDs traditionally are considered diseases of adults, many of them can start in childhood and are outlined in Table 409.2.
TABLE 409.2. CLASSIFICATION OF THE LIMB-GIRDLE MUSCULAR DYSTROPHIES (LGMD) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


