Henoch-Schönlein Syndrome
Leslie M. Higuchi
Robert P. Sundel
Henoch-Schönlein syndrome (HSP) is the most common vasculitis in children, occurring in 20 children per 100,000 per year. It is a multisystem disorder that principally involves the skin, gastrointestinal tract, joints, and kidneys. Heberden is credited with describing the first case in 1801. Other names for the syndrome include anaphylactoid, rheumatoid or allergic purpura, leukocytoclastic vasculitis, and Henoch-Schönlein purpura. Most cases of HSP occur in children younger than 7 years, with a peak age range between 4 and 7 years. In adults, an inflammatory vasculopathy sharing features with HSP is typically diagnosed as hypersensitivity vasculitis, although this is increasingly thought to be the same condition. The male-to-female ratio is 1.5–2:1. Peak seasonal incidence has been reported in the fall, winter, and spring.
ETIOLOGY
Although the etiology of HSP remains unknown, several predisposing factors have been suggested. Upper respiratory infections precede the onset of illness in at least 50% of children with the syndrome. Streptococcal infections are most common, although numerous others have been reported including parvovirus, adenovirus, Mycoplasma pneumoniae, Yersinia, Legionella, Epstein-Barr virus, and varicella. Insect bites, exposure to cold, and vaccination against measles, cholera, yellow fever, typhoid, and paratyphoid A and B preceded HSP in other cases.
All the conditions known to incite HSP are far more common than the vasculitis itself, so additional factors related to host susceptibility also must play a role in the condition. Reports of the familial occurrence of HSP suggest a genetic connection, but because families also may have similar environmental exposures, such evidence is only circumstantial. Additional support for an inherited predisposition comes from children transplanted for end-stage renal failure associated with HSP. In one report, nine histologic renal recurrences of HSP occurred among 12 living related donor grafts, but no recurrences developed in the five cadaveric kidneys. The specific genetic loci involved are not known, although HSP has been associated with genes of the major histocompatibility complex (MHC) located on chromosome 6, including a weak relationship to HLA-B35 and HLA-DR4, as well as aberrant expression of the fourth component (C4) of the complement system.
PATHOPHYSIOLOGY
HSP is thought to be an immune-mediated vasculitic disorder. Abnormalities in patients include the deposition of immunoglobulin A (IgA) in affected organs and elevated serum IgA concentrations. In vitro studies of lymphocytes from patients with HSP demonstrate an increased number of IgA-bearing and -secreting B cells, with altered T-cell regulation of antibody synthesis. Both IgA and IgG immune complexes and IgA rheumatoid factor have been described, and unconfirmed reports also have pointed to a possible contribution of autoantibodies of the IgA isotype. IgA is able to activate the alternative pathway of complement; and C3, properdin, and membrane attack complexes (C5 through C9) may be demonstrated within the glomerular mesangium of children with HSP. In addition, total serum hemolytic complement (CH50) and properdin levels may be depressed, and levels of C3d, a C3 breakdown product, may be elevated. Thus, plasma proteins may form aggregates that activate the alternative pathway of the complement system, thus leading to deposition within target organs, release of a cascade of inflammatory mediators and, eventually, tissue injury.
The centrality of any of these findings is, however, controversial. The IgA “complexes” actually may represent IgA aggregates or IgA complexed to complement-fixing proteins such as IgG or fibronectin, and many patients have no evidence of autoantibodies. Recently, therefore, some authors have suggested that abnormal O-glycosylation of IgA leads to deposition in the renal mesangium and development of HSP. Alternatively, some evidence points to a role for factor XIII in the pathogenesis of HSP. The disease is characterized by bleeding into affected organs, despite a normal platelet count and normal prothrombin and partial thromboplastin times. Factor XIII, also known as fibrin-stabilizing factor, is decreased in these patients. According to this alternate hypothesis, proteases released during the local tissue inflammatory response may degrade factor XIII, resulting in tissue fibrin deposition and contributing to the development of vasculitis. Interestingly, the gene encoding the factor XIII A chain, containing the catalytic function, is found on chromosome 6, near MHC genes potentially associated with HSP.
Whatever the trigger, histopathologic features of HSP are typical of other leukocytoclastic vasculitides. Small vessels have
a perivascular infiltrate consisting of neutrophils and mononuclear cells. Deposits of IgA, C3, and fibrin may be detected using immunofluorescence and electron microscopy. Renal biopsies typically show an endocapillary proliferative glomerulonephritis involving endothelial and mesangial cells, with deposition of IgA, C3, fibrin or fibrinogen, properdin, IgG and, less commonly, IgM. Proliferation of extracapillary cells also may occur, resulting in variable degrees of crescent formation.
a perivascular infiltrate consisting of neutrophils and mononuclear cells. Deposits of IgA, C3, and fibrin may be detected using immunofluorescence and electron microscopy. Renal biopsies typically show an endocapillary proliferative glomerulonephritis involving endothelial and mesangial cells, with deposition of IgA, C3, fibrin or fibrinogen, properdin, IgG and, less commonly, IgM. Proliferation of extracapillary cells also may occur, resulting in variable degrees of crescent formation.
CLINICAL MANIFESTATIONS AND COMPLICATIONS
The typical clinical presentation is that of a previously well child who develops malaise and low-grade fever followed by palpable purpura, colicky abdominal pain, and arthritis. The manifestations may occur simultaneously or sequentially over a period of several days or weeks. About half of the children with HSP present with a skin rash (Fig. 436.1). The classic eruption of HSP begins as localized or generalized urticarial wheals that are replaced by erythematous macules or maculopapules. Subsequently, petechiae and larger purpuric areas form. Often, the purpuric lesions are palpable and described as raised papules or plaques that, similar to the stages of ecchymoses, evolve over several days from red-purple to yellow and then to purple-brown before fading. The lesions usually arise in crops, and new crops may arise at different times, resulting in a polymorphic appearance with varying stages of eruption simultaneously present.
Typically, the rash is nonpruritic and favors pressure-dependent areas, with a characteristic distribution on the lower extremities and the buttocks in toddlers and older children. Younger patients who are not ambulatory may show involvement of the upper extremities, face, and trunk. The rash may be transient or persist for weeks, and it may recur. In addition to the classic rash, various forms of erythema multiforme, with central hemorrhage or ulceration, vesicular eruptions, and bullae, have been described. Additionally, nonpitting edema occurs in 35% to 70% of patients, more commonly in children younger than 3 years. Affected areas are tender and distorted, with localized swelling having a predilection for the scalp, ears, periorbital region, and the dorsum of the hands and feet. More diffuse swelling of dependent areas and the periorbital regions also may occur as a secondary complication of nephrosis.
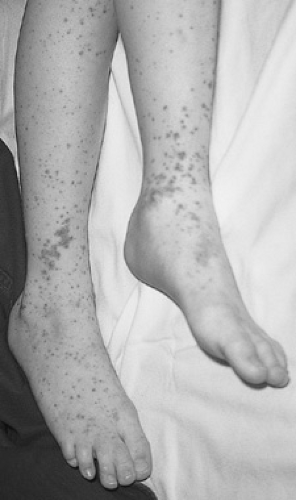 FIGURE 436.1. This patient demonstrates the classic appearance of the rash over the lower distal extremity. (Reproduced with permission from Fleisher GR, Ludwig S, Baskin MN. Atlas of pediatric emergency medicine. Philadelphia: Lippincott Williams & Wilkins, 2004:311.) |
The second clinical feature that makes up the diagnostic triad of HSP is joint involvement. Arthralgia or arthritis develops in 65% to 85% of patients and is the presenting symptom or sign of the illness, preceding the rash, in 17% to 25% of patients. The joint involvement is nonmigratory, and both articular and periarticular tissues are affected. Whereas joints may be extremely painful, erythema and warmth are uncommon. Joint involvement more commonly affects the knees and ankles than the smaller joints of the fingers and wrists and usually is self-limited, never resulting in residual sequelae.
The final cardinal manifestation of HSP, gastrointestinal involvement, occurs in as many as three-fourths of patients. The colicky abdominal pain frequently causes tenderness, but rebound is uncommon. Gastrointestinal complaints may precede the rash in 15% of cases, creating particular difficulty in differentiating the condition from appendicitis or other abdominal pathology. Often, gastrointestinal bleeding occurs, manifesting as melena or guaiac-positive stools (56%) or hematemesis (10%). Massive gastrointestinal hemorrhage (5%) is rare. Vomiting and ileus are reported in 25% and 40% of cases, respectively.
The typical abdominal pain of HSP is intermittent or waxing and waning. An acute change in the nature, intensity, or pattern of abdominal pain is often a sign of secondary intestinal complications such as intussusception, bowel infarction, perforation, pancreatitis, or hydrops of the gallbladder. Of these, intussusception is the most common, occurring in 3% of patients. It is more common in older children, and is ileoileal in 65% of cases, in contrast, to sporadic cases of intussusception which tend to be ileocolic and frequently occur in children younger than 3 years. Other rare gastrointestinal complications include protein-losing enteropathy and late-onset ileal stricture formation. Hepatomegaly, of uncertain significance, is noted in 10% of patients.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


