General Approaches to Coagulopathies
Clifford M. Takemoto
James F. Casella
INTRODUCTION
Abnormalities of the hemostatic system are encountered commonly in hospitalized children, because primary diseases of hemostasis often require hospitalization, and systemic diseases severe enough to require hospitalization often produce abnormalities of hemostasis. In addition, pediatricians are frequently called on to evaluate coagulation abnormalities before or after surgery.
Coagulation disorders can be divided into conditions with abnormal bleeding (i.e., hypocoagulable states) and those associated with the development of thromboses (i.e., hypercoagulable states). Current knowledge of factors necessary for normal clotting to occur and for maintaining blood in a fluid state is sufficiently complex that a full understanding of all the mechanisms involved is a challenge even for an experienced hematologist. New information is being acquired rapidly, especially in the area of hypercoagulability. Despite this surge of new information, most abnormalities of hemostasis can still be approached in an orderly fashion, beginning with a detailed history and physical examination, the evaluation of readily available laboratory tests, and a general overview of hemostatic mechanisms.
Coagulopathies may be secondary to abnormalities of the blood vessels, platelets, or plasma clotting factors, and some hemostatic defects involve more than one of these systems. The result of the history, including the family pedigree, physical examination, and screening laboratory testing, should provide information about which system is responsible for the bleeding abnormality. The following paragraphs summarize the basic functions of each component of the hemostatic mechanism and provide a background for understanding the mechanisms underlying clinical abnormalities. The relative contributions of the history, physical examination, and laboratory testing are then discussed.
NORMAL PHYSIOLOGY
Coagulation Mechanisms
The primary response to bleeding includes a vascular response, platelet activation, and the coagulation cascade. Within seconds of injury, damaged blood vessels demonstrate a vasoconstrictive response. During this vasoconstriction phase, platelets begin to adhere to the damaged endothelium, where collagen, a potent activator of platelet adhesion and von Willebrand factor (vWF) ligand, are exposed. Adherence and aggregation of platelets appear to be mediated through the action of specific platelet membrane receptors, including glycoprotein Ib (the major vWF receptor) and glycoprotein IIb/IIIa (the fibrinogen receptor). Adherent platelets then release platelet-stimulatory agents, such as thromboxane and adenosine diphosphate, and mediators of platelet interaction, such as fibrinogen and vWF, promoting adherence and activation of more platelets, with the resulting formation of an unstable hemostatic plug. Serotonin released from platelet granules further constricts the large vessels. Platelet factor 4, a substance released from platelet granules, neutralizes heparin. Receptors for activated factors V (FVa) and VIII (FVIIIa) are also expressed on the phospholipid membranes of activated platelets.
Coincident with these events, the coagulation system is activated, leading to the activation of thrombin and the subsequent deposition of an insoluble fibrin clot. This system has traditionally been modeled as a linear cascade of events activated at two levels (“extrinsic and intrinsic”) (Fig. 296.1) resulting in clot formation; however, the emerging picture is one of an interacting set of enzyme complexes that coordinate the initiation, amplification, and dissolution of the fibrin clot. These complexes consist of serine proteases and cofactors that are activated by proteolysis and require calcium and a phospholipid surface for full activity. The initial activation of this system in vivo begins through the extrinsic pathway with the exposure of tissue factor (TF) from damaged tissue. TF forms a complex with the serine protease, activated factor VII (FVIIa). This complex (TF/FVIIa), also called the extrinsic tenase, then activates factor X (FX) in the presence of calcium and phospholipid on the surface of cell membranes. Activated FX (FXa) assembles with its cofactor, FVa on the surface of activated platelets to form the prothrombinase complex. This prothrombinase complex, in turn, cleaves prothrombin to thrombin, and thrombin then cleaves soluble fibrinogen to fibrin, which polymerizes into a fibrin gel. In addition, thrombin further potentiates clotting by activating both platelets and factor XIII (FXIII), a trans-glutaminase that stabilizes the clot by covalently cross-linking fibrin.
Thus, clot formation in vivo initiates through the TF/FVIIa complex and the extrinsic pathway. However, TF/FVIIa can also activate the intrinsic pathway to a lesser degree by cleaving factor IX (FIX) to activated FIX (FIXa). The intrinsic pathway is important for the propagation of the clot by a positive feedback loop in which thrombin activates components of the intrinsic system (FIX, FVIII, and FV) and thus amplifies thrombin production. FVIIIa and FVa combine to form the intrinsic tenase complex that cleaves FX on a phospholipid surface in the presence of calcium. The distinction of the “intrinsic” versus the “extrinsic” system is useful in understanding the components that affect the prothrombin time (PT) and activated partial thromboplastin time (aPTT). Deficiencies of factors of the extrinsic system result in PT prolongation, whereas deficiencies of factors of the intrinsic system lead to aPTT elevation. However, PT and aPTT measurements do not faithfully predict physiologic bleeding. Patients with deficiencies of factor XII, (FXII) prekallikrein, or high-molecular-weight kininogen have prolongation of the aPTT, but no bleeding symptoms. Similarly, soluble factors, such as a lupus anticoagulant, can prolong
the aPTT, but they may infer a risk of thrombosis rather than hemorrhage. FXII and factor XI (FXI), prekallikrein, and high-molecular-weight kininogen can be activated on negatively charged surfaces, and together they comprise what is termed the contact activation pathway of coagulation.
the aPTT, but they may infer a risk of thrombosis rather than hemorrhage. FXII and factor XI (FXI), prekallikrein, and high-molecular-weight kininogen can be activated on negatively charged surfaces, and together they comprise what is termed the contact activation pathway of coagulation.
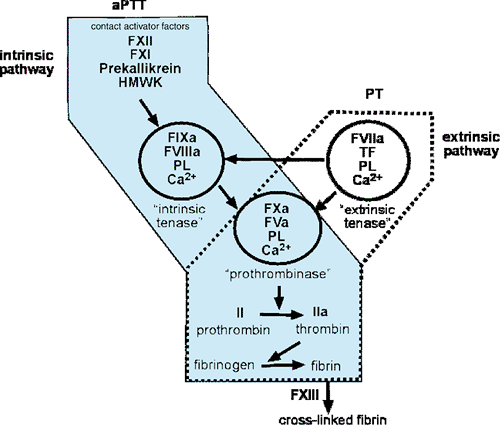 FIGURE 296.1. The coagulation cascade. The major enzyme systems that regulate the intrinsic and extrinsic pathway are depicted. The “prothombinase” complex contains activated factors X and V (FXa and FVa) and requires calcium (Ca2+) and a phospholipid surface (PL). The prothrombinase complex activates prothrombin (II) to thrombin (IIa), which subsequently converts soluble fibrinogen to the fibrin clot. Factor XIII (FXIII) cross-links fibrin monomers to stabilize the clot. The prothrombinase complex is activated in two ways, by either the “intrinsic” tenase (FIXa and FVIIIa) or the “extrinsic” tenase (activated factor VII [FVIIa] and tissue factor [TF]). The intrinsic tenase can be activated by FVIIa/TF and by the contact activation factors (factor XII (FXII), factor XI (FXI), prekallikrein, and high-molecular-weight kininogen [HMWK]). The factors that are known to affect the activated partial thromboplastin time (aPTT) are within the shaded area, whereas the factors that are known to affect the prothrombin time (PT), are contained within the area outlined with the dotted lines. |
Anticoagulation Mechanisms
The presence of such an elegant system for the formation of blood clots implies that an equally sophisticated system must be in place to prevent the inadvertent formation of blood clots and to provide for their removal and remodeling once formed. Three major systems inhibit the coagulation system: (a) the protein C/S system, (b) antithrombin, and (c) the TF pathway inhibitor (TFPI). In addition, removal of the thrombus through fibrinolysis is mediated by the plasminogen system. Protein C, an anticoagulant protein, plays a primary role in inhibiting the clotting cascade and inducing the dissolution of clots. Like some of the procoagulant factors (e.g., factor II [FII], FVII, FIX, and FX), protein C is a vitamin K–dependent factor synthesized in the liver. Unlike the procoagulant factors, activated protein C possesses anticoagulant activity, which is achieved primarily through cleavage and inactivation of FVa and FVIIIa. Activated protein C also stimulates fibrinolysis.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


