Fig. 13.1
Univariate correlation between ectopic fat and organ-specific insulin resistance
HGP, hepatic glucose production; FPI, fasting plasma insulin; SSPI, steady state plasma insulin; IHL, intrahepatic lipid; IMCL, intramyocellular lipid; VO2, basal oxygen consumption rate per body weight. *p < 0.05, **p < 0.01, ***p < 0.001 (Modified from Ref. [11])
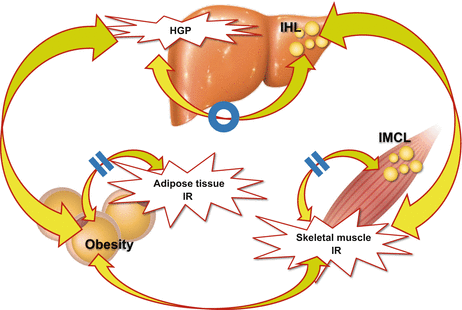
Fig. 13.2
Relationship between intracellular lipid accumulation and organ-specific insulin resistance among three major insulin-targeting organs, the liver, skeletal muscle and adipose tissue
Skeletal muscle insulin resistance is not associated with intramyocellular lipid accumulation, but with hepatic steatosis, suggesting that there is inter-organ network between the liver and skeletal muscle in human (Modified from Ref. [11])
13.3 Possible Molecular Mechanisms Underlying Mystery of the Selective Insulin Resistance in the Type 2 Diabetic Liver
In the liver, insulin suppresses glucose production and enhances lipogenesis. Indeed, total insulin resistance in the liver observed in the LIRKO mice present hyperglycemia without fatty liver [8]. However, in the liver with type 2 diabetes, insulin fails to suppress gluconeogenesis but still activates lipogenesis, the pathology of which is regarded as a ‘selective insulin resistance’-like phonotype [18] (Fig. 13.3). Notably, the indices for hepatic fat accumulation are associated with HGP × FPI, but not with % suppression of HGP during the hyperinsulinemic clamp (Fig. 13.1) [11], suggesting that the liver fat is associated with basal HGP itself, possibly independently of insulin resistance. Indeed, HGP is not solely regulated by insulin, but also by other hormones such as glucagon and glucocorticoid. Transcription factors such as forkhead box protein O (FoxO), cAMP response element-binding protein (CREB), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), hepatocyte nuclear factor 4α (HNF4α) and glucocorticoid receptors are involved in the regulation of gluconeogenesis by the hormones and nutrients. In addition, hepatic steatosis also occurs independently of insulin resistance. Of the triacylglycerol accumulated in the liver of patients with NAFLD, 59.0 % arises from non-esterified fatty acids mainly derived from the adipose tissue and 14.9 % from the diet, whereas only 26.1 % from de novo lipogenesis [19]. Transcription factors such as liver X receptor (LXR), sterol regulatory element-binding protein-1c (SREBP-1c) and carbohydrate response element binding protein (ChREBP) are involved in the de novo lipogenesis. Of these, SREBP-1c is a master regulator of hepatic de novo synthesis of fatty acids and is activated not only by insulin but also by glucose, fatty acids, amino acids, SREBP-1c itself, LXR, mechanistic target of rapamycin complex 1 (mTORC1) and endoplasmic reticulum (ER) stress [20].
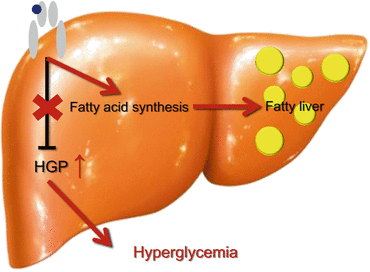
Fig. 13.3
Selective insulin resistance-like phenotype in the liver with type 2 diabetes
In the liver with type 2 diabetes, insulin fails to suppress gluconeogenesis but continues to activate lipogenesis, which is regarded as a ‘selective insulin resistance’-like phonotype
Triglyceride itself is not a toxic lipid and rather may be protective to prevent toxic effects of free fatty acids. Rather, free cholesterol and saturated free fatty acid palmitate are regarded as toxic lipids that cause hepatic insulin resistance via oxidative stress [17] as follows:
1.
Mice fed an atherogenic diet rich in cholesterol and cholate show steatohepatitis, oxidative stress and hepatic insulin resistance [4]. In the liver of this model, SREBP-1c is upregulated possibly via cholesterol-mediated LXR activation, leading to de novo lipogenesis in the liver. In addition, hepatic expression of insulin receptor substrate (IRS)-2 was downregulated by cholesterol diet. Shimano et al. discovered that SREBP-1c binds to E-box in the promoter of IRS-2 competitively with TFE3 and FoxO1, and thus downregulates IRS-2 [21]. These findings may link lipogenesis and insulin resistance in the liver.
2.
In an in vitro fatty liver system using H4IIEC3 hepatocytes, a saturated fatty acid palmitate, but not an unsaturated fatty acid oleate, inhibits insulin-stimulated tyrosine phosphorylation of insulin receptor substrate 2 and serine phosphorylation of Akt, through c-Jun NH2-terminal kinase (JNK) activation [5]. In this model, mitochondrial β oxidation-derived reactive oxygen species (ROS) play a causal role in the palmitate-induced JNK activation [5]. Therefore, toxic lipid-induced mitochondrial ROS may also underlie the link between steatosis and insulin resistance in the liver. Indeed, in human, genes involved in mitochondrial oxidative phosphorylation (OXPHOS) are coordinately upregulated and positively correlated with those involved in a ROS-related pathway in the livers of obese type 2 diabetic patients compared with those of non-obese type 2 diabetic patients [7]. These findings in toxic lipids-induced hepatic steatosis also contribute to the selective insulin resistance-like phonotype in type 2 diabetic livers [18].
13.4 Crosstalk Among Glucose, Protein and Lipid Metabolic Pathways in the Liver
Growing evidence suggests that chronic endoplasmic reticulum (ER) stress in the liver is a major contributor to obesity-induced insulin resistance [22, 23]. ER is responsible for protein quality control [24]. The burden of unfolded proteins in the ER lumen is identified as an ER stress by several ER stress sensors, known as ATF6, PERK and IRE-1. The ER stress sensors trigger cellular adaptation for unfolded protein accumulation to restore normal function of the cell, which is called as the unfolded protein response (UPR). Chronic UPRs are causally linked to the pathogenesis of human metabolic diseases such as obesity and type 2 diabetes. Accumulating evidence suggests that obesity promotes ER stress, which is detected as an enhanced UPR signaling, that activates JNK and impairs insulin signaling at the level of IRSs in the liver and adipose tissue [25]. However, the molecular mechanisms linking obesity and ER stress were not fully understood.
In searching for metabolic pathways that are significantly altered by obesity in the livers of people with type 2 diabetes, we found that genes involved in ubiquitin-proteasome pathways are coordinately upregulated in obese individuals [7]. Proteasomes play fundamental roles in processes that are essential for cell viability by degrading misfolded proteins [26]. Unexpectedly from the expression data, liver proteasome activity was reduced by approximately 30–40 % in mouse models of obesity, such as genetically obese ob/ob mice, diabetic db/db mice and C57BL/6 mice fed a high fat diet (HFD) [27]. As a consequence, ubiquitinated proteins were accumulated in the liver of these obese model mice. These results suggest that liver proteasome activity is reduced in animal models of obesity. Thus, coordinate upregulation of the genes involved in the ubiquitin-proteasome pathway in obese patients and mouse models of obesity may compensate for impaired proteasome function. Therefore, we hypothesized that proteasome dysregulation in the liver is involved in the development of hepatic insulin resistance in obesity and type 2 diabetes. To test this hypothesis, we generated PA28α-PA28β-PA28γ triple-knockout (PA28 KO) mice, the genes of which are up-regulated in the livers of patients with obesity and in those of mice fed HFD [27], as a model of impaired proteasome function and investigated their metabolic phenotypes as follows: (1) Hepatic proteasome activity in PA28 KO mice fed a standard chow is reduced by 35 % as compared with wild-type mice. As expected, ubiquitinated proteins are accumulated in the liver of the PA28 KO mice. (2) Electron micrographs reveals massive expansion of the ER in the livers of PA28 KO mice, suggestive of UPR. Indeed, the liver in PA28 KO mice show the evidence of ER stress, such as increased levels of Grp78, CHOP, p-PERK, p-eIF2α and p-IRE-1α, as well as ER stress-inducible mRNAs encoding CHOP and the spliced form of XBP-1 (XBP-1s), as compared with wild-type mice. (3) Phosphorylation of JNK and its downstream target c-Jun are significantly increased in the livers of PA28 KO mice as compared with those of wild-type mice. (4) Although body weight is not altered, PA28 KO mice fed the standard chow present glucose intolerance. Hyperinsulinemic-euglycemic clamp experiments and western blot analysis of the insulin signaling pathway reveal that PA28 deficiency impairs insulin signaling mainly in the liver, but not in the skeletal muscle, and thereby induces systemic glucose intolerance in vivo. These findings illustrate that proteasome dysfunction causes ER stress, JNK activation and thereby cause insulin resistance in the liver (Fig. 13.4). Yang et al. reported that hepatic autophagy is downregulated in the livers of ob/ob mice and that defective autophagy in Atg7 KO mice causes ER stress and hepatic insulin resistance [28]. Therefore, it is possible that both proteasome- and autophagy-mediated protein degradation are impaired in the livers of obese individuals, further exacerbating ER stress.
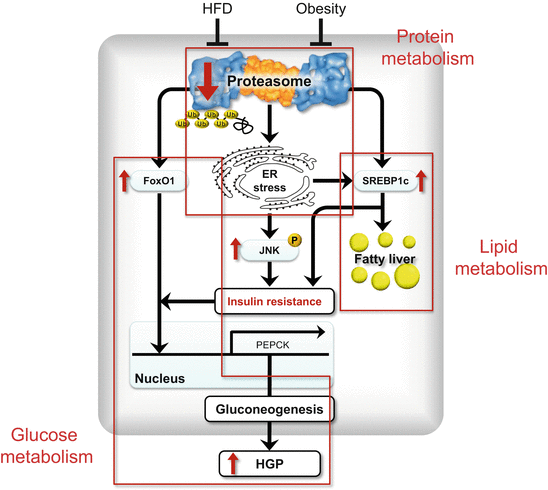
Fig. 13.4
Crosstalk among protein, glucose and lipid metabolic pathways
Proteasome function is impaired in the state of obesity, followed by endoplasmic reticulum (ER) stress, JNK activation and insulin resistance in the liver. Proteasome dysfunction also results in increased protein levels of FoxO1 and SREBP-1c because proteasome is responsible for the degradation of FoxO1 and SREBP-1c, which are master transcription factors for gluconeogenesis and lipogenesis, respectively. SREBP-1c is also activated by ER stress. Such activation of these factors occurs independently of insulin resistance and may mimic so-called ‘selective insulin resistance’ in the liver with type 2 diabetes, that is, coexistence of fatty liver and enhanced gluconeogenesis (Modified from Ref. [27])
(5) PA28 KO mice show hepatic steatosis associated with upregulated Srebf1 and Acc1 and increased cleaved/active SREBP-1c (Fig. 13.4). SREBP-1c is activated by ER stress [20]. In addition, proteasome dysfunction results in increased protein levels of SREBP-1c because proteasome is responsible for the degradation of SREBP-1c [29].
(6) FoxO1 protein amounts dramatically increase in both cytoplasmic and nuclear fractions, probably due to proteasome dysfunction in the liver of PA28 KO mice (Fig. 13.4). Spliced XBP-1 directly binds FoxO1 and promotes its protein degradation via the proteasome [30]. In the liver of PA28 KO mice, spliced XBP1 protein is increased, probably due to increased phosphorylation of IRE1α, an endonuclease for XBP1 gene. In addition, hepatic insulin resistance caused by ER stress/JNK pathway and increased SREBP-1c that downregulates IRS-2 further accumulates FoxO1 in the nucleus, leading to induction of genes involved in gluconeogenesis such as Pepck1.
These findings suggest that proteasome dysfunction may be a primary event linking obesity and ER stress-induced insulin resistance in the liver. In addition, there seems to be a crosstalk among protein-, glucose- and lipid-metabolism pathways (Fig. 13.3). Notably, the activation of SREBP-1c and FoxO1 occurred independently of insulin resistance, and may mimic so-called ‘selective insulin resistance’ in the liver with type 2 diabetes, that is, coexistence of fatty liver and enhanced gluconeogenesis (Fig. 13.2).
Proteasome function seems to be altered differently in different tissues. Insulinopenic hyperglycemia impairs proteasome activity in the liver and kidney [31, 32], whereas proteasome activity is enhanced in the wasted muscle of obese diabetic db/db mice [33]. Taken together, these findings indicate that obesity predominantly induces proteasome dysfunction in the liver. This clarifies the previous finding that ER stress causes insulin resistance in the liver together with the adipose tissue [22] and brain [34]. Significance of enhanced proteasome activity in the skeletal muscle of obese model mice should be investigated in future.
13.5 Role of Hepatokines that Mediate Inter-Organ Network During Remodeling of Energy Homeostasis
As described above, we hypothesized that a liver-derived hormone, hepatokine, affects the distant organ insulin sensitivity. Human hepatic gene expression information accumulated by using serial analysis of gene expression (SAGE) technique and DNA chip methods [35, 36] were used to identify genes with signal peptides whose hepatic expression levels were significantly correlated with glycemic control (HbA1c), obesity (BMI) or insulin resistance (HOMA-R and metabolic clearance rate). Expression of the candidates hepatokine genes were further referred to the various animal models of diabetes, obesity and fatty liver [4, 37–39]. Based on these approaches, we isolated 62 candidate genes for hepatokines associated with insulin resistance, hyperglycemia and obesity [35].
13.5.1 Selenoprotein P
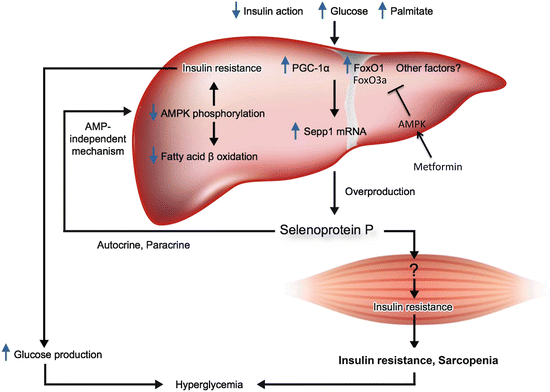
Of these, we identified a gene encoding selenoprotein P, the expression levels of which were positively correlated with insulin resistance and hyperglycemia [40]. Indeed, serum levels of selenoprotein P are elevated in people with type 2 diabetes and significantly correlated with fasting plasma glucose and HbA1c levels [40]. Selenoprotein P (in humans encoded by the SEPP1 gene) is upregulated through FoxOs (Fig. 13.5). Insulin downregulates SEPP1 by phosphorylating and inactivating FoxO1 [41], whereas antidiabetic metformin activates AMP-activated protein kinase (AMPK), phosphorylates and inactivates FoxO3a, and thereby downregulates SEPP1 in hepatocytes [42].
 < div class='tao-gold-member'>
< div class='tao-gold-member'>
 Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
 Mechanism of Skeletal Muscle Contraction: Role of Mechanical Muscle Contraction in Glucose Homeostasis
Mechanism of Skeletal Muscle Contraction: Role of Mechanical Muscle Contraction in Glucose Homeostasis
 Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
 Fracture Risk in Diabetes
Fracture Risk in Diabetes
 Body Temperature Regulation During Exercise Training
Body Temperature Regulation During Exercise Training




Only gold members can continue reading. Log In or Register to continue
Related posts:
Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
Mechanism of Skeletal Muscle Contraction: Role of Mechanical Muscle Contraction in Glucose Homeostasis
Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
Fracture Risk in Diabetes
Body Temperature Regulation During Exercise Training
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
