Fig. 12.1
Overview of carbohydrate and lipid metabolism. Glycolysis is utilized by all tissues for the catabolism of glucose to provide energy in the form of ATP. Pyruvate dehydrogenase irreversibly converts pyruvate, the end product of glycolysis, into acetyl-CoA. This compound is a primary fuel for the TCA cycle and the platform for FA synthesis. The major function of the TCA cycle is the oxidation of acetyl-CoA to CO2 and H2O. In this process, NAD+ is reduced to NADH, resulting in the generation of ATP. Glucose is stored as glycogen in an easily metabolizable form. Glycogen is rapidly degraded in contracting muscle. Excess amounts of glucose are converted to FAs through acetyl-CoA and acyl-CoA. Dietary FA enters cells and forms FA-CoA. When needed as fuel, FA is subjected to β oxidation in mitochondria. For storage, FA is esterified as TG and then incorporated into lipid droplets. When energy is needed, TG is reversely degraded into FA and glycerol. Glucose 6-P glucose 6-phosphate, FA fatty acid, FA-CoA fatty acyl-CoA, MG monoacylglycerol, DG diacylglycerol, TG triacylglycerol, TCA cycle tricarboxylic acid cycle, NAD+ nicotinamide adenine dinucleotide, IMCL intramyocellular lipid
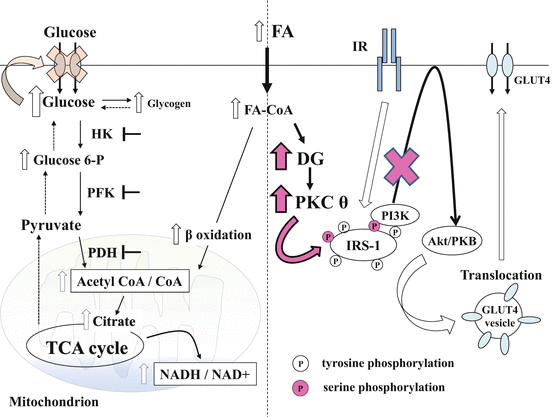
Fig. 12.2
FA-induced insulin resistance (hypothesis and revision). Intracellular FA influx causes β oxidation, resulting in increased ratios of acetyl-CoA to CoA and NADH to NAD+ in mitochondria. As a result, PDH is inactivated. Simultaneously, the accumulation of citrate inhibits PFK and the subsequent increase of glucose 6-P levels, inhibition of HK, and promotion of glycogen synthesis. Therefore, intracellular glucose accumulation inhibits glucose uptake (left side). However, recent findings suggest that both intracellular glucose 6-P and glucose levels are decreased in FA-induced insulin-resistant skeletal muscle. Alternatively, it has been hypothesized that FA-induced impairment of insulin stimulates glucose uptake. Increased transport of FA transiently increases DG content, leading to PKC-θ activation. Stimulated PKC-θ induces serine phosphorylation in IRS-1, which antagonizes the insulin-stimulated tyrosine phosphorylation in IRS-1. Thus, the binding of IRS-1 to PI3K is blocked, resulting in the inhibition of downstream insulin signaling, including the final step of GLUT4 translocation (right side). Glucose 6-P glucose 6-phosphate, HK hexokinase, PFK phosphofructokinase, PDH pyruvate dehydrogenase, TCA cycle tricarboxylic acid cycle, NAD+ nicotinamide adenine dinucleotide, FA fatty acid, FA-CoA fatty acyl-CoA, DG diacylglycerol, TG triacylglycerol, PKC-θ protein kinase C θ, IR insulin receptor, IRS-1 insulin receptor substrate-1, PI3K phosphatidylinositol 3 kinase, GLUT4 glucose transporter 4
Concerning lipids, diet-originated FAs are transported in the circulation as either TG in lipoprotein and hydrolyzed by lipoprotein lipase or bound to albumin [2]. After uptake into cells, a CoA moiety is ligated to FA by acyl-CoA synthase. Fatty acyl-CoA enters mitochondria for oxidation or the TG synthesis pathway for energy storage, especially in adipocytes (Fig. 12.1). Against energy demand, FAs from TG in lipid droplets are catalyzed by three lipases. Adipose triglyceride lipase, hormone-sensitive lipase, and monoacylglycerol lipase degrade TG, diacylglycerol (DG), and monoacylglycerol, respectively. Finally, the generated FAs and glycerol are released into the bloodstream for transport to other tissues (Fig. 12.1) [2, 3].
12.3 Intramyocellular Lipid Metabolism and Insulin Resistance
12.3.1 Insulin Resistance and Ectopic Fat Accumulation in Skeletal Muscle
As opposed to adipose tissue, which can be visualized as obesity under high calorie intake, it has been difficult to quantify IMCLs. Through muscle biopsy, it has been reported that TG content of skeletal muscle is usually less than 0.5 % volume density in a lean subject and can increase to more than 1.5 % in obese individuals [4]. However, proton nuclear magnetic resonance spectroscopy (1H-MRS) has made it possible to noninvasively measure IMCLs consisting primarily of TG [5]. Recent findings suggest that IMCLs are strongly associated with insulin resistance in skeletal muscle [6, 7]. Diet- and exercise-induced reduction of IMCL levels resulted in improvement of muscle insulin resistance [8]. Therefore, IMCLs appear to be a causal factor for obesity-related insulin resistance. However, trained athletes with high insulin sensitivity display relatively high levels of IMCLs. This phenomenon is known as ‘the athlete’s paradox’ [9], as we will discuss later. To solve this mystery, both IMCLs (intramyocellular fat accumulation) and extracellular and/or intracellular lipid (FAs) should be considered. The complicated interactions among these factors could induce insulin resistance as a consequence. Insulin resistance is defined as a failure of insulin-stimulated glucose uptake into skeletal muscle through the glucose transporter 4 (GLUT4). Therefore, we attempt to discuss the mechanism by which either an extracellular or intracellular lipid could interrupt intracellular insulin signaling, considering the following connection with intracellular lipids (IMCLs).
12.3.2 Extramyocellular FAs and Insulin Resistance
12.3.2.1 The Glucose-FA Cycle Described by Randle et al.
It has long been known that FAs can induce insulin resistance. Randle et al. proposed a hypothesis considering the link between intracellular glucose and lipid metabolism [10]. High concentrations of extracellular FAs could increase its influx into cells. Consequently, enhanced β oxidation of FAs occurs in mitochondria, resulting in increased ratios of acetyl-CoA to CoA and NADH to NAD+ (Fig. 12.2: left side). As lipid oxidation (increase of acetyl-CoA content) provides a sufficient energy supply for the TCA cycle, PDH is inactivated, and glucose oxidation is reduced. The accumulation of citrate resulting from the increased β oxidation of FAs inhibits PFK, which is one of the rate-limiting enzymes of glycolysis. Then, an increase of glucose 6-phosphate (glucose 6-P) levels results in additional negative feedback and HK inhibition. Therefore, an increase in intracellular glucose levels could inhibit insulin-stimulated glucose uptake as a negative feedback mechanism of energy balance (Fig. 12.2: left side). This hypothesis is attractive and apparently theoretical. In this case, increased intracellular concentrations of glucose and glucose 6-P as well as glycogen are expected in skeletal muscle. However, it has been extremely difficult to prove this hypothesis in humans without repeated muscle biopsies. Even if a sample can be obtained, short-term hypoxia induces changes in metabolite concentrations.
12.3.2.2 Revision of the Traditional Hypothesis
To overcome these difficulties, Shulman’s group adopted carbon-13 (13C) and phosphorus-31 (31P) MRS. This state-of-the-art technique has enabled non-invasive measurements of intramyocellular G6P, glycogen, and glucose levels. Insulin-stimulated glucose uptake and intracellular glucose metabolism were evaluated in the presence of high FA levels under a euglycemic hyperinsulinemic clamp using 13C-MRS and 31P-MRS combined with muscle biopsy samples. Surprisingly, reductions in intramyocellular glucose 6-P and glucose concentrations and decreased glycogen synthesis were observed, suggesting that insulin-stimulated glucose transport activity was inhibited by FA [11]. Therefore, the traditional hypothesis that impaired insulin-stimulated glucose uptake due to negative feedback of FA-induced inhibition of glycolysis has been revised [12, 13].
The next question is at what point is insulin signaling inhibited in FA-induced insulin resistance. Although the detailed mechanism of insulin-stimulated glucose uptake via GLUT4 translocation to the plasma membrane is unclear, FAs appear to inhibit a relatively proximal step of insulin signaling [14]. Examination using muscle biopsy samples unveiled reduced insulin receptor substrate-1 (IRS-1) – associated phosphatidylinositol 3-kinase (PI3K) activity as described in subsequent sections.
12.3.3 Intramyocellular Lipid and Insulin Resistance
Excess circulating FAs are taken up into skeletal muscle by passive diffusion and protein-mediated transport [15]. Excess intramyocellular FAs are oxidized or esterified as TG and then incorporated into lipid droplets for storage. As described previously, because IMCL content is strongly associated with insulin resistance, it appears to partly explain obesity-related insulin resistance in skeletal muscle. However, based on this concept, ‘the athlete’s paradox’ is not fully understood [16].
12.3.3.1 IMCL and Diacylglycerol Acyltransferase 1 (DGAT1)
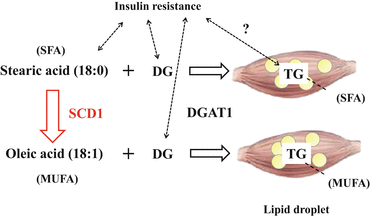
One of the key factors for explaining this discrepancy may be DGAT1, which transfers FAs from fatty acyl-CoA to DG to create TG (Fig. 12.3). In other words, DG but not TG appears capable of playing a central role in insulin resistance. For example, mice overexpressing DGAT1 in skeletal muscle display a dissociation of DG and TG content. These mice accumulate TG with low DG content in skeletal muscle, but they are protected against fat-induced insulin resistance in skeletal muscle [17]. In addition, a single session of exercise increased muscle DGAT1 expression accompanied by increased TG, reduced DG, and improved FA-induced insulin resistance in humans. In turn, activation of the pro-inflammatory JNK/NF-κB pathway was suppressed regardless of IMCL accumulation [18]. These findings suggest that DG but not TG can cause insulin resistance. TG may be metabolically ‘inactive.’
Focusing on the function of DGAT1 in skeletal muscle, it could act as a protector against insulin resistance. On the contrary, DGAT1-null mice are resistant to diet-induced obesity and insulin resistance. Interestingly, transplantation of adipose tissue from DGAT1-null mice to wild-type mice results in decreased adiposity and increased insulin sensitivity [19]. Contrary to skeletal muscle, SCD1 deficiency in adipose tissue may be favorable. We must always consider the organ being discussed and the links among organs.
12.3.3.2 IMCLs and Stearoyl-CoA Desaturase-1 (SCD1)
Another factor may be stearoyl-CoA desaturase-1 (SCD1), which converts saturated fatty acids (SFAs) into monounsaturated fatty acids (MUFAs). SCD1 introduces cis-double bonds between the 9th and 10th carbons of acyl-CoA, mainly in stearoyl-CoA (18:0) or palmitoyl-CoA (16:0) to generate oleoyl-CoA (18:1 (9)) or palmitoleoyl-CoA (16:1 (9)), respectively [20, 21] (Fig. 12.3). Regarding the SCD1 and SCD5 isoforms in humans, SCD1 is highly expressed in skeletal muscle [21]. It is known that SFAs, such as stearic acid (18:0), are generally toxic. In comparison, MUFAs, such as oleic acid (18:1), are cytoprotective [22]. Similarly, SFAs can more readily induce insulin resistance compared to MUFAs [23]. Moreover, SCD1 contributes to lipid bilayer fluidity by adjusting the appropriate ratio of MUFAs/SFAs in the cell membrane through the conversion of SFAs to MUFAs [21].
Thus, it is expected that higher SCD1 expression could be beneficial with regard to metabolic disorders. In fact, both acute bouts of aerobic exercise [18] and chronic endurance exercise [4] can increase SCD1 expression in skeletal muscle. SCD1 expression is higher in athletes than that in sedentary subjects [4], although both groups display increased IMCL levels. Elevated SCD1 may promote increased MUFA content in IMCLs, which may result in higher lipid quality. Again, SCD1 may explain ‘the athlete’s paradox’ [21].
However, as previously noted in DGAT1-null mice, whole-body SCD1-deficient mice adversely exhibit enhanced energy expenditure and improved insulin signaling [24, 25]. Unfortunately, data from skeletal muscle-specific SCD1 deficient mice are not available at present. Regardless, further studies will be needed to clarify the role of SCD1 in skeletal muscle.
 < div class='tao-gold-member'>
< div class='tao-gold-member'>
 Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
 Mechanism of Skeletal Muscle Contraction: Role of Mechanical Muscle Contraction in Glucose Homeostasis
Mechanism of Skeletal Muscle Contraction: Role of Mechanical Muscle Contraction in Glucose Homeostasis
 Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
 Fracture Risk in Diabetes
Fracture Risk in Diabetes
 Body Temperature Regulation During Exercise Training
Body Temperature Regulation During Exercise Training




Only gold members can continue reading. Log In or Register to continue
Related posts:
Bone Disease Associated with Diabetes Mellitus: Particularly Focusing on Its Contribution to the Development of Atherosclerosis
Mechanism of Skeletal Muscle Contraction: Intracellular Signaling in Skeletal Muscle Contraction
Mechanism of Skeletal Muscle Contraction: Role of Mechanical Muscle Contraction in Glucose Homeostasis
Ectopic Fat Accumulation and Glucose Homeostasis: Role of Leptin in Glucose and Lipid Metabolism and Mass Maintenance in Skeletal Muscle
Fracture Risk in Diabetes
Body Temperature Regulation During Exercise Training
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
