Disorders of Transport
Rebecca S. Wappner
An increasing number of inborn errors of metabolism are being recognized to result from defects in the transport of various types of metabolites or compounds across cell or organelle membranes. Most often the defect involves a specific carrier mechanism, often protein-based, which is subject to genetic mutation. This chapter discusses the classic defects involving the renal transport of amino acids and that of phosphate. Examples of other defects in transport, discussed elsewhere in this section, include lysinuria protein intolerance and hyperammonemia–hyperornithinemia–homocitrullinuria, discussed with disorders of the urea cycle; microsomal glucose transporter defects associated with glycogen storage diseases types Ib and Ic; and defective lysosomal transport of free sialic acid in Salla disease and infantile free sialic acid storage disease, cystine in cystinosis, and vitamin B12 in combined homocystinuria and methylmalonic acidemia.
DISORDERS OF RENAL AMINO ACID TRANSPORT
Cystinuria
Cystinuria is named after the extremely elevated urinary cystine concentration found in affected persons. The very low solubility of cystine leads to its precipitation and recurrent stone formation in the genitourinary tract. Affected patients may present at any age with acute abdominal or flank pain (colic), hematuria, urinary tract infection, renal failure, or hydronephrosis or other obstruction of the urinary tract. The disorder may be accompanied by a history of passing small stones known as “gravel” or crystals in diapers. Occasionally, presymptomatic patients will be diagnosed by the finding of characteristic hexagonal cystine crystals on routine urinalysis.
The disorder results from defects in the low-specificity, high-capacity renal tubular reabsorption transport system for the dibasic amino acids (ornithine, arginine, and lysine) and cystine. Fig. 384.1 shows the similar structure of four amino acids involved; the amino groups are approximately the same distance apart. Lysine is excreted in much larger amounts than is cystine but, because it is fairly soluble, it does not form stones. Intestinal transport of dibasic amino acids also is affected but is not of clinical significance.
Cystinuria occurs in approximately 1 in 7,000 persons. The disorder results from mutations at two separate genetic loci, SLC3A1 on chromosome 2 and SLC7A9 on chromosome 19, which code for the heavy and light chains, respectively, of the renal cystine/dibasic amino acid transport system. Clinically, two types of the disorder are recognized, which are distinguished by the urine cystine excretion pattern in carriers and by the mode of inheritance. Carriers for type I cystinuria have normal urinary excretion of cystine. Type I cystinuria, inherited on an autosomal recessive basis, is most often associated with mutations at the SLC3A1 locus, but can be seen with SLC7A9 mutations. Carriers for nontype I cystinuria (previously termed type II and type III) have moderately increased urinary cystine levels, even to the extent that occasionally they form stones. This pattern of inheritance is most consistent with an autosomal dominant mode with incomplete penetrance. Nontype I
cystinuria most often results from mutations at the SLC7A9 locus. Affected persons with type I, nontype I, or heterozygous combined type I/nontype I cystinuria cannot be distinguished by clinical findings or urine cystine levels. Type I cystinuria and nontype I cystinuria are of approximately equal incidence.
cystinuria most often results from mutations at the SLC7A9 locus. Affected persons with type I, nontype I, or heterozygous combined type I/nontype I cystinuria cannot be distinguished by clinical findings or urine cystine levels. Type I cystinuria and nontype I cystinuria are of approximately equal incidence.
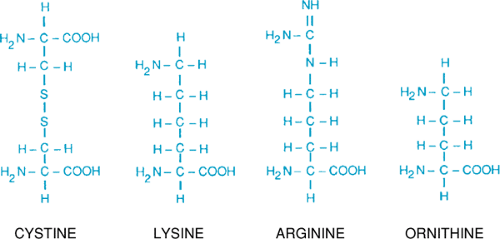 FIGURE 384.1. The four amino acids excreted in excess in cystinuria. |
The diagnosis may be suggested by analysis of excreted stones. Quantitative measurement of amino acids in a timed urine sample will document the disorder in patients older than 6 months. Before that time, immaturity of renal tubular transport frequently interferes with the ability to distinguish homozygotes from heterozygotes for nontype I cystinuria. Usually, urinary cystine excretion of more than 250 mg/g of creatinine is diagnostic of cystinuria.
Management is directed at preventing stone formation. Oral intake of liquids is increased to keep the urinary cystine concentration to less than 300 mg/L. Alkalinization of the urine to a pH of more than 7.5 will increase the solubility of cystine. If recurrent stones form, the use of D-penicillamine or one of its homologues, 2-mercaptopropionylglycine, may be indicated. These compounds form a mixed disulfide with the cysteinyl residues of cystine that are soluble and excreted readily. Renal transplantation may be needed for advanced renal disease.
Iminoglycinuria
Iminoglycinuria, a benign disorder, results from defects in another low-specificity, high-capacity transport system of the renal tubule that is shared by the two imino acids—proline and hydroxyproline—and glycine. The diagnosis is confirmed by quantitative urine amino acid measurements. Plasma concentrations of all three acids will be normal or low.
Other disorders resulting in increased plasma proline or hydroxyproline concentrations also may cause iminoglycinuria in that the specific imino acid involved will so overload the renal tubular reabsorption system as to interfere with reabsorption of the other imino acid and that of glycine. Transient iminoglycinuria may be seen also in normal neonates.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


