Disorders of the Liver and Biliary System
Donald A. Novak
Frederick J. Suchy
William F. Balistreri
Significant changes in hepatic anatomy and physiology occur in neonates. These rapid maturational alterations are required for infants to cope with their changing environment.
THE LIVER AND GALLBLADDER IN EARLY LIFE
Postnatally, the liver grows in proportion to infants’ height, weight, and age; in addition, the proportions of the liver change. The liver may account for as much as 10% of the total body volume in fetuses, but constitutes approximately 5% of body weight at birth and only 2% of adult weight.
Clinical Assessment of Liver Size
The standard clinical indices of liver size are the degree of projection of the liver edge below the right costal margin, the span of dullness on percussion, and the length of the vertical axis of the liver, estimated radiographically. Several studies have provided a baseline of age-related values. In 1957, McNicholl’s study of 317 healthy infants and children established values for the projection of the liver edge below the costal margin and emphasized that a projection of greater than 3.5 cm in the midclavicular line (MCL) in the newborn indicated hepatic enlargement. The measurement of liver span may be subject to less variability than is the degree of projection. Younoszai and Mueller measured liver span (vertical height) in older patients by percussing the upper margin and palpating the lower edge in the right MCL. A linear increase in the span, similar in both genders, correlated with body weight and age. Lawson et al. determined the liver span in the MCL in 350 infants and children by percussion of the upper and lower borders. The mean liver span was found to be curvilinearly related to age; by extrapolation, the range was estimated to be 1.9 cm at 1 week of life, to 7.7 cm (for men) and 6.3 cm (for women) at age 20. Age and gender were the major factors correlating liver size in children with normal growth patterns. The expected span of liver dullness in the MCL in children ages 12 to 20 can be obtained by the following equations:
Boys: span (cm) = [0.032 × weight (lb)]
+ [0.18 × height (in.)] 7.86
Girls: span (cm) = [0.027 × weight (lb)]
+ [0.22 × height (in.)] 10.75
+ [0.18 × height (in.)] 7.86
Girls: span (cm) = [0.027 × weight (lb)]
+ [0.22 × height (in.)] 10.75
Reiff and Osborn determined that the mean liver span in 100 healthy newborns was 5.9 ± 0.8 cm by percussion; this value, much larger than the one cited earlier, suggests that extrapolation of the curve by Lawson et al. may not be accurate for the neonatal age range. It exhibited a poor correlation of span with measurement of liver projection below the costal margin. Reiff and Osborn emphasized the clinical utility of the assessment of liver span.
Liver volume may reflect liver size more accurately because a change in shape may not necessarily be distributed equally. Liver volume, as assessed by ultrasonography, a reproducible and noninvasive method, correlated inversely (r = -0.79) with age. The relative volume (expressed per unit of body weight) in the first year of life (approximately 48 mL/kg) was almost twice the volume at age 15 (approximately 25 mL/kg).
Clinical Assessment of Gallbladder Size
The gallbladder is visualized readily in children, even in neonates, using high-frequency real-time ultrasonographic imaging. A gallbladder that may be distended in sick infants, often in conjunction with sepsis, mandates essential definitions of those criteria useful in differentiating a normal gallbladder in a newborn from a gallbladder or biliary tract that is pathologically enlarged or atretic. In a study of children between ages 1 month and 16 years, ultrasonography scans showed a gradual age-related increase in the size of the gallbladder. Wall thickness never was more than 3 mm; the lumen of the common hepatic duct increased with age but never was greater than 4 mm.
Functional Alterations during Development
Specific deficiencies in hepatic function have been observed repeatedly in normal, healthy newborn infants. This finding has led to an extensive investigation of the alterations in the quantitative pattern of various enzymes during embryonic development. Despite marked intraspecies differences, several general concepts can be stated.
During late fetal and early postnatal development, the differentiation of tissue function depends on de novo synthesis of enzymes, not on activation of enzymes already present in the embryonic tissue. Greengard documented the quantitative pattern of enzymatic differentiation in early life and observed that the increase in their concentrations (i.e., emergence) occurs in clusters that correlate with the changing functional requirements of the developing organism. Substrate and hormonal flow across the placenta and dietary and hormonal input in the postnatal period modulate the emergence of these enzymatic processes. The lipid composition of various cell and organelle membranes changes rapidly in response to alterations in the composition of dietary lipids. The role of genetic preprogramming (i.e., effect of the biologic clock) also must be considered in understanding the ontogeny of metabolic activities.
Age-Related Differences in Standard Biochemical Assays
Well-defined, age-related changes are seen in the biochemical parameters of hepatic function. Normal reference values are cited in Appendix A. For example, serum ceruloplasmin and alpha1-antitrypsin values do not vary significantly with age after the immediate perinatal period. Kattwinkel et al. determined that serum levels of aspartate aminotransferase, gamma-glutamyltransferase, 5′-nucleotidase, and total alkaline phosphatase and its bone isoenzymes exhibit significant age dependency in normal children. Throughout early life, cholesterol concentrations increase rapidly. Importantly, clinicians at each institution must establish a working range using their instrumentation.
Alkaline Phosphatase
The total activity of alkaline phosphatase in human serum varies considerably with age and, to some degree, with gender. The largest proportion of these age-dependent differences is caused by an increased isoenzyme originating in bone; these fluctuations are therefore most significant during periods of accelerated bone growth. Values must be interpreted in the context of these known physiologic changes. In newborns, the reference normal values may be as much as four times the values of adults; throughout the remainder of childhood, alkaline phosphatase activity may be three times the adult value, with a decline after puberty.
Ammonia
Except for a transient increase in the ammonia content of peripheral blood in normal neonates, ammonia nitrogen values during early life are similar to adult values. Transient hyperammonemia is seen in early life, possibly due to the shunting of blood through the ductus venosus and immaturity of metabolic processes.
Alpha-Fetoprotein
The concentrations of alpha-fetoprotein (αFP), a glycoprotein synthesized by embryonic tissues (e.g., liver, yolk sac), are highest in serum during the tenth to fourteenth weeks of fetal life. Concentrations decrease rapidly, especially in the perinatal period.
Gamma-Glutamyltransferase (γ-GTP)
Gamma-glutamyltransferase (gamma-glutamyl transpeptidase) is a multifunctional, membrane-bound glycoprotein that serves catalytic and detoxification functions. Because of the difficulty in interpreting alkaline phosphatase values, the measurement of gamma-glutamyl transpeptidase may be beneficial in the pediatric population; activity is normally high at birth and declines rapidly with maturation.
5′-Nucleotidase
Usually, serum 5′-nucleotidase activity is elevated in hepatobiliary diseases. However, unlike serum alkaline phosphatase, its activity is not increased in infancy, childhood, skeletal disorders, or pregnancy.
CONGENITAL ANOMALIES AND ABERRATIONS OF HEPATIC PHYSIOLOGY IN EARLY LIFE
Hepatic structural and functional abnormalities due to congenital or acquired defects can manifest in the perinatal period. The response of the neonatal liver to injury from a variety of insults may be stereotypic: the formation of giant cells, inflammation, and cell necrosis.
Anomalies
Reductions or increases in the external lobation of the liver are rare. More frequent are abnormalities of the hepatic ducts and gallbladder, such as partial or complete duplication or congenital absence of the gallbladder. Riedel’s lobe, a tongue-shaped mass of normal liver tissue that projects downward from the right lobe, is a common anatomic variation.
Hepatomegaly
In the evaluation of unexplained hepatomegaly (Box 367.1), ultrasonography can demonstrate hepatic size and consistency. A hyperechogenic (bright) appearance of the hepatic parenchyma is common in children and can be caused by metabolic disease (e.g., glycogen storage disease) or a fatty liver (e.g., obesity, malnutrition, hyperalimentation, steroid therapy). This ultrasonographic finding in children can guide further evaluation, such as liver biopsy with quantitative and qualitative assays for fat or specific enzyme assays.
Chronic Cholestasis
Infants and children with hepatobiliary dysfunction, regardless of the cause, are at risk for the sequelae of prolonged cholestasis. These sequelae include malabsorption of fats and fat-soluble vitamins, secondary to diminished intestinal bile acid concentrations, and retention of endogenous compounds, such as bilirubin and bile acids, usually excreted by the liver. Chronic liver disease, with resultant impairment of hepatic function, may alter nutrient metabolism. Growth failure may result from aberrations in hormonal balance. Hepatic disease may progress, with the eventual development of hepatic cirrhosis, portal hypertension, and liver failure. Therapy is largely symptomatic; in most cases, progression of the underlying hepatic disease cannot be halted. General recommendations for therapy are given in Table 367.1.
Patients with chronic cholestasis may develop intense pruritus, manifest in early life. The cause of pruritus remains obscure; xanthomas also may develop. Xanthoma formation may be related to the elevated serum lipid values found in chronic cholestasis. Therapy for both complications is aimed at inducing more efficient excretion of these endogenous compounds. Ursodeoxycholic acid (10 to 30 mg/kg/day in divided doses) is the agent of choice. Cholestyramine resin (8 to 16 g/day) also may be given to stimulate bile excretion. The usefulness of this agent is limited by the extent of residual bile flow. In addition, cholestyramine is unpalatable, its administration is difficult, and it may cause constipation and fat-soluble vitamin deficiency. Rifampin (10 mg/kg/day; maximum, 300 mg/day) has been evaluated in a few pediatric patients with pruritus and has been found efficacious, as has partial biliary diversion. Therapy with ultraviolet B (UVB) light has proven useful in the control of pruritus in adults. Plasma perfusion and therapy using carbamazepine or hydroxyzine have been used in adults, but their usefulness in children must be evaluated. Newer therapies have focused on the central causes of pruritus. Specifically, naltrexone, an opiate receptor antagonist, and ondansetron, acting via 5-hydroxytryptamine receptors, have been shown to be efficacious in relieving the pruritus associated with cholestasis in adults.
Often, nutritional inadequacies are a problem in children with chronic cholestasis. Fat malabsorption is common, because of diminished intestinal bile acid concentrations. It is
essential to provide adequate calories and supplements containing medium-chain triglycerides (which are absorbed more readily) as orally administered formula or through nasogastric drip. Fat-soluble vitamin (e.g., A, D, E, K) deficiency is common in children with chronic cholestasis. Vitamin D deficiency may cause rickets; vitamin K deficiency may be responsible for intracranial hemorrhage; vitamin E deficiency may be responsible for a progressive neuromuscular syndrome once thought to be an inherent feature of multiple cholestatic diseases. A deficiency (i.e., malabsorption) of vitamin E causes a neuropathy characterized by progressive loss of the myelinated axons of peripheral nerves and a degeneration of spinal cord posterior columns; the neuronal tocopherol content may be low. Clinically, areflexia, cerebellar ataxia, ophthalmoplegia, and peripheral neuropathies are seen. Because these lesions appear to be partly reversible in young children, careful monitoring of fat-soluble vitamin levels is necessary. Vitamin E levels may be elevated falsely because of elevated serum lipid levels, and the ratio of serum vitamin E to total serum lipids (normal ratio, less than 0.6 mg/g in children younger than 12 years) should be followed. Oral supplementation should be given as necessary, but intramuscular administration has been required in some patients. D-Alpha-tocopheryl polyethylene glycol-1,000 succinate (TPGS) is a water-soluble form of vitamin E that is adsorbed well after oral administration and is safe and effective for the prevention and correction of the vitamin E deficiency of chronic cholestasis.
essential to provide adequate calories and supplements containing medium-chain triglycerides (which are absorbed more readily) as orally administered formula or through nasogastric drip. Fat-soluble vitamin (e.g., A, D, E, K) deficiency is common in children with chronic cholestasis. Vitamin D deficiency may cause rickets; vitamin K deficiency may be responsible for intracranial hemorrhage; vitamin E deficiency may be responsible for a progressive neuromuscular syndrome once thought to be an inherent feature of multiple cholestatic diseases. A deficiency (i.e., malabsorption) of vitamin E causes a neuropathy characterized by progressive loss of the myelinated axons of peripheral nerves and a degeneration of spinal cord posterior columns; the neuronal tocopherol content may be low. Clinically, areflexia, cerebellar ataxia, ophthalmoplegia, and peripheral neuropathies are seen. Because these lesions appear to be partly reversible in young children, careful monitoring of fat-soluble vitamin levels is necessary. Vitamin E levels may be elevated falsely because of elevated serum lipid levels, and the ratio of serum vitamin E to total serum lipids (normal ratio, less than 0.6 mg/g in children younger than 12 years) should be followed. Oral supplementation should be given as necessary, but intramuscular administration has been required in some patients. D-Alpha-tocopheryl polyethylene glycol-1,000 succinate (TPGS) is a water-soluble form of vitamin E that is adsorbed well after oral administration and is safe and effective for the prevention and correction of the vitamin E deficiency of chronic cholestasis.
BOX 367.1 Pathophysiology and Differential Diagnosis of Liver Enlargement
Increased Number of Cells in the Liver
Inflammation (hepatocyte or Kupffer cell enlargement, inflammatory cells)
Viral, acute or chronic
Bacterial (sepsis, abscess, cholangitis)
Toxic
Autoimmune
Storage
Fat
Nonalcoholic fatty liver disease
Reye syndrome/mitochondrial disease
Malnutrition
Lipid infusion
Cystic fibrosis
Diabetes
Glycogen (multiple forms of glycogen storage disease)
Specific lipid storage disease
Gaucher disease
Niemann-Pick disease
Wolman disease
Miscellaneous
Alpha one antitrypsin deficiency
Wilson disease
Hypervitaminosis A
Infiltration
Primary tumors
Hepatoblastoma
Hepatocellular carcinoma
Hemangioma
Focal nodular hyperplasia
Secondary or metastatic tumors
Lymphoma
Leukemia
Neuroblastoma
Wilms tumor
Hemophagocytic lymphohistiocytosis
Increased Size of Vascular Space
Intrahepatic obstruction to hepatic vein outflow
Venoocclusive disease
Hepatic vein thrombosis (Budd-Chiari)
Hepatic vein web
Suprahepatic
Congestive heart failure
Pericardial disease
Tamponade
Constrictive pericarditis
Increased Size of Biliary Space
Congenital hepatic fibrosis
Caroli disease
Idiopathic (benign?)
Despite careful attention to caloric intake, mineral balance, and vitamin status, poor hepatic synthetic function may limit growth. At this point, affected patients may become candidates for orthotopic liver transplantation (OLT). Survival rates up to 90% are reported following OLT. The success rates and degree of organ availability increase significantly as infants attain adequate size (larger than 10 kg), although hepatic size–reduction techniques have made transplantation more available to smaller children, as has the use of living related donors.
Total Parenteral Nutrition–Related Cholestasis
Parenteral nutrition is a life-saving form of therapy in neonates, children, or adults who are unable to receive enteral nutrition. However, serious complications are frequent, among which is cholestasis related to total parenteral nutrition (TPN), second in incidence only to catheter-related sepsis.
Cholestasis associated with TPN occurs primarily in premature infants, although hepatobiliary lesions, including triaditis and steatosis, have been described in older children and adults. In an early report, the incidence of cholestasis in children receiving TPN was 50% in infants with birth weights of less than 1,000 g and 18% in infants with birth weights of 1,000 to 1,500 g. Other investigators have confirmed the inverse relation between gestational age and the incidence of TPN-related cholestasis. The onset of cholestasis appears to be related to the duration of infusion, usually occurring after at least 2 weeks of therapy. Frequently, TPN-related hepatic disease occurs in sick, premature infants, typically those undergoing episodes of sepsis, shock, abdominal surgery, and necrotizing enterocolitis.
TPN-related cholestasis has an insidious onset. Jaundice may be observed, but it is attributed to “physiologic” hyperbilirubinemia. Often, the diagnosis of TPN-related cholestasis is entertained first when routine parenteral nutrition–related surveillance laboratory tests reveal elevated serum levels of conjugated bilirubin. Usually, serum bile acid levels are elevated; often, an increase in serum bile acid levels is the earliest biochemical abnormality associated with TPN-related cholestasis. Later in the disease course, serum aminotransferase and serum alkaline phosphatase levels may become abnormal. Typically, hepatic synthetic function remains normal until late in the disease course.
The differential diagnosis of TPN-related cholestasis includes the many causes of neonatal cholestasis and is a diagnosis of exclusion. The evaluation should concentrate on identifying other potentially treatable causes of cholestasis, including infectious, metabolic, or anatomic disorders that may be detected in as many as 10% of infants evaluated for possible TPN-related liver dysfunction. This exercise is imperative, because alternative therapy may be available.
TABLE 367.1. SUGGESTED MEDICAL MANAGEMENT OF THE CONSEQUENCES OF PERSISTENT CHOLESTASIS | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
The hepatic pathology of TPN-related cholestasis is nonspecific. Early changes include hepatocyte and canalicular cholestasis, extramedullary hematopoiesis, giant cell transformation, and pseudoacinar formation. Later changes reflect worsening disease, with inflammatory infiltrates, ductular proliferation, and fibrosis. Although hepatic fibrosis, cirrhosis, and hepatic failure have been documented in patients receiving long-term TPN, milder changes, including ductular proliferation and inflammation, can reverse upon discontinuation of TPN and resumption of oral feedings.
The cause of TPN-related cholestasis is unknown. In early life, affected infants have a period during which hepatic uptake and (presumably) the excretion of bile acids and other organic anions is diminished. Premature infants may be unusually susceptible to cholestasis. Elevated serum concentrations of the potentially toxic bile acid lithocholate occur during prolonged TPN administration and may potentiate cholestasis. Fasting alone may predispose affected infants to cholestasis through lack of the hormonal stimulation of bile secretion. Sepsis, common in the infant receiving TPN, is known to cause cholestasis. Repeated episodes of intravascular catheter infection seem to be associated with worsening disease, especially in patients with short bowel syndrome. Amino acids have been implicated in the cause of TPN-associated cholestasis, and indirect evidence suggests that amino acid infusions may influence the development of cholestasis. Certain amino acids have been shown to inhibit hepatic bile acid uptake. The amino acid composition of TPN infusates is capable of altering bile flow rates in animals. Unfortunately, a specific amino acid deficiency or toxicity syndrome resulting in cholestasis has not been identified in the human. Choline deficiency is common in patients on long-term TPN, and may result in hepatic steatosis, as may excessive rates of glucose infusion. Little evidence is available to suggest that intravenous fat or glucose infusions are associated with cholestasis. Other possible factors in the genesis of TPN-related cholestasis include deficiencies or excesses of various minerals or trace elements.
The management of patients with TPN-related cholestasis is difficult. Continuation of TPN may result in worsening hepatic disease. Conversely, complete withdrawal of TPN may result in a catabolic state and poor growth. A prudent approach is to attempt the slow introduction of enteral feeding, to stimulate gastrointestinal hormone release and bile flow. The protein infusion should be limited to the lowest amount required for growth. Avoiding episodes of sepsis, through scrupulous care of indwelling catheters, as well as of bacterial translocation, through the use of enteral feedings, with resultant improvement in mucosal integrity may be beneficial. The use of intermittent courses of oral antibiotics also has been advocated, especially in those patients with short bowel syndrome who suffer from bacterial overgrowth. The use of minimal enteral feedings through the oral administration of approximately 10% of the daily caloric needs may prevent or ameliorate TPN-related cholestasis.
Generally, the prognosis for infants who develop TPN-related cholestasis is good. However, long-term follow-up studies are not available.
TPN-related cholelithiasis occurs primarily in preterm infants. Patients may be asymptomatic or may present with signs and symptoms of acute cholecystitis, including vomiting, rapid
rise of serum bilirubin, fever, and sepsis. Diagnosis depends on clinical suspicion and ultrasonography. Factors important in the genesis of TPN-related cholestasis also may play a role in the causation of cholelithiasis; in particular, fasting may be associated with gallbladder stasis. Continued fasting and TPN administration may cause biliary sludge, composed of thick bile intermixed with pigment granules, calcium bilirubinate, and cholesterol crystals. Sludge, identifiable by ultrasonography, appears to be the precursor of stone formation, and prolonged gallbladder stasis may predispose to stone formation. The administration of furosemide and of TPN solutions may increase calcium secretion into bile, increasing its lithogenicity. Gallbladder sludge resolves spontaneously with the resumption of enteral feedings. Rarely, gallstones have been reported to clear with feeding. Cycling of TPN administration does not appear to be helpful.
rise of serum bilirubin, fever, and sepsis. Diagnosis depends on clinical suspicion and ultrasonography. Factors important in the genesis of TPN-related cholestasis also may play a role in the causation of cholelithiasis; in particular, fasting may be associated with gallbladder stasis. Continued fasting and TPN administration may cause biliary sludge, composed of thick bile intermixed with pigment granules, calcium bilirubinate, and cholesterol crystals. Sludge, identifiable by ultrasonography, appears to be the precursor of stone formation, and prolonged gallbladder stasis may predispose to stone formation. The administration of furosemide and of TPN solutions may increase calcium secretion into bile, increasing its lithogenicity. Gallbladder sludge resolves spontaneously with the resumption of enteral feedings. Rarely, gallstones have been reported to clear with feeding. Cycling of TPN administration does not appear to be helpful.
Total Parenteral Nutrition–Associated Hepatobiliary Dysfunction in Older Patients
Although hepatic dysfunction with TPN is reported most often in infants, some reports describe the development of cholestasis—associated with bile ductular proliferation, periportal inflammation, and fibrosis—in older patients who have undergone massive intestinal resection, requiring prolonged parenteral nutrition. Less severe abnormalities, including steatosis without cholestasis, have been observed in adults with GI dysfunction requiring TPN. It is unclear whether these cases share a pathogenesis similar to that of TPN-related cholestasis of infancy.
METABOLIC DISEASE OF THE LIVER
The liver plays a central role in carbohydrate, lipid, and amino acid synthesis and degradation. Not surprisingly, the liver is involved primarily or secondarily in many states of metabolic derangement. For example, the absence of a crucial enzyme may cause a build-up of toxic metabolites; this condition is found in patients with tyrosinemia. Conversely, the sequestration of a synthesized product within the liver may lead to hepatic and systemic damage, as is seen in alpha1-antitrypsin deficiency.
Hepatic metabolic disease may be suggested by a family history or by the pattern of symptom onset. For example, liver injury after the initiation of fructose ingestion should suggest the diagnosis of fructosemia. Clinical features of liver-based metabolic diseases are nonspecific but include jaundice, hepatosplenomegaly, failure to thrive, dysmorphism, developmental delay, hypotonia, seizures, and progressive neuromuscular dysfunction. Screening laboratory data may reveal hypoglycemia, hyperammonemia, increased serum aminotransaminase levels, acidosis, or hypoprothrombinemia. The metabolic disease can be confirmed in a variety of ways. Percutaneous hepatic biopsy allows histologic examination and measurement of specific enzyme activities or substrate accumulation. Accurate diagnosis is important in that it may allow effective therapy and genetic counseling. Therefore, physicians must remain alert to the possibility of metabolic disease.
Disorders of Tyrosine Metabolism
Tyrosine is an amino acid important in the synthesis of melanin, thyroid hormones, and catecholamines. Metabolism of tyrosine to fumaric acid and acetoacetic acid proceeds down a pathway with several intermediates. Deficiency or immaturity of any of the enzymes catalyzing these steps may lead to hypertyrosinemia. Transient neonatal tyrosinemia is a self-limiting condition of premature neonates, presumably caused by an immaturity of tyrosine aminotransferase activity. Vitamin C may be effective in enhancing enzyme activity in these patients. Hypertyrosinemia may occur also in any form of severe hepatic injury, typically in concert with high serum methionine levels.
Hereditary Tyrosinemia
Type 1 hereditary tyrosinemia is an autosomal recessive disorder. The acute form manifests in infancy with symptoms of jaundice, failure to thrive, anorexia, hepatosplenomegaly, ascites, hypoprothrombinemia, clinical bleeding, rickets, and hepatic failure or cirrhosis. Usually, the chronic form is seen later in life, accompanying cirrhosis or hepatocellular carcinoma. Episodic, severe, acute peripheral neuropathy is common in patients surviving infancy and is an important cause of morbidity and mortality.
Laboratory features include diminished hepatic synthetic function, including decreased vitamin K–dependent clotting factors and hypoalbuminemia. Serum aminotransferase levels are mildly to moderately elevated, as is serum bilirubin. Hemolytic anemia or hypoglycemia may occur. Fanconi syndrome, with attendant hyperphosphaturia, glycosuria, proteinuria, and aminoaciduria, often is evident. Rickets, presumably secondary to hypophosphatemia, may complicate the picture. Serum tyrosine values are extraordinarily high, as may be serum methionine levels. The phenolic acid by-products of tyrosine metabolism (i.e., p-hydroxyphenyl lactic, p-hydroxyphenyl pyruvic, and p-hydroxyphenyl acetic acids) are excreted in the urine, as are succinyl acetone and succinyl acetoacetate.
Pathogenesis
The enzymatic defect in type 1 tyrosinemia appears to be at the level of fumarylacetoacetate (FAH). The gene responsible for this activity has been mapped to human chromosome 15, and multiple mutations have been associated with the phenotype of type 1 tyrosinemia. This deficiency causes a build-up of potentially toxic metabolites, including succinyl acetone (SA) and succinyl acetoacetate (SAA). These intermediates are toxic reactive metabolites that may cause renal and hepatic damage as a result of binding to sulfhydryl groups of proteins.
Hepatic Pathology
The hepatic injury in tyrosinemia presumably begins in utero, as illustrated by elevated cord blood αFP levels with normal serum tyrosine levels. The liver exhibits nodular cirrhosis and extensive fibrosis. Pseudoacinar formation, fatty infiltration, and iron deposition may occur. Hepatocellular carcinoma may be found in older patients with cirrhosis.
Therapy
The acute form of type 1 tyrosinemia, without therapy, typically is fatal in the first year of life. Dietary modifications to limit the intake of phenylalanine, tyrosine, and methionine appear to improve renal dysfunction, but the effects on the hepatic disease are unclear. Proper diagnosis is important for genetic counseling, and prenatal diagnosis is available. The treatment of choice for infants with type 1 tyrosinemia is 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione, an inhibitor of 4-hydroxyphenylpyruvate dioxygenase. Further study is required to determine the role of this agent in preventing both ongoing hepatic damage and subsequent malignant transformation; however, current evidence suggest that this agent, when begun early in life, may significantly diminish the severity of hepatic disease as well as the incidence of hepatocellular carcinoma in those afflicted with tyrosinemia. Liver transplantation can correct most of the biochemical
abnormalities of hereditary tyrosinemia and, therefore, has been recommended for all tyrosinemic children with cirrhotic nodules demonstrated on computed tomography (CT) or ultrasonographic examinations, because of the great risk of developing hepatocellular carcinoma. Neurologic crises provide an additional reason to consider early liver transplantation.
abnormalities of hereditary tyrosinemia and, therefore, has been recommended for all tyrosinemic children with cirrhotic nodules demonstrated on computed tomography (CT) or ultrasonographic examinations, because of the great risk of developing hepatocellular carcinoma. Neurologic crises provide an additional reason to consider early liver transplantation.
(Disorders of carbohydrate and lipoprotein metabolism and their effect on the liver are discussed in Chapters 387 and 388.)
DISORDERS OF PROTEIN METABOLISM: UREA CYCLE ENZYME DEFECTS
Hyperammonemia
The differential diagnosis of childhood hyperammonemia includes disorders of the urea cycle, mitochondrial disorders, organic acidemias, disorders of fatty acid oxidation, transient hyperammonemia of the neonate, Reye syndrome, drug toxicity (e.g., secondary to valproate), and hepatic failure. Differentiation of these situations is important because specific therapy is available for certain disorders.
Ammonia is eliminated from the body through the formation of urea in the liver (Fig. 367.1). An accumulation of this potentially toxic substrate can be ascribed to primary or acquired alterations in any of the enzymatic steps in this cycle. Episodes of hyperammonemia may depress consciousness and eventually may produce permanent impairment of neurologic function, or death. Symptoms in newborns may be more subtle.
Most often, hyperammonemia in newborns is secondary to inherited defects in urea cycle enzymes. If untreated, deficiency in ornithine transcarbamylase, a sex-linked recessive disorder, is fatal in the neonatal period. Affected heterozygous girls also may have episodic hyperammonemia, with subsequent mental retardation or death. Citrullinemia and argininosuccinic acidemia may manifest in the neonatal period with hyperammonemic coma or, in a subacute form with mental retardation; those with argininosuccinate lyase deficiency may undergo progressive hepatic fibrosis. Type II citrullinemia has been associated with idiopathic neonatal hepatitis. Patients with argininemia have hyperammonemia and spastic diplegia. Affected newborns may have a clinical picture resembling that caused by sepsis. A high index of suspicion is required. An algorithm for the workup of infants with hyperammonemia is shown in Figure 367.2.
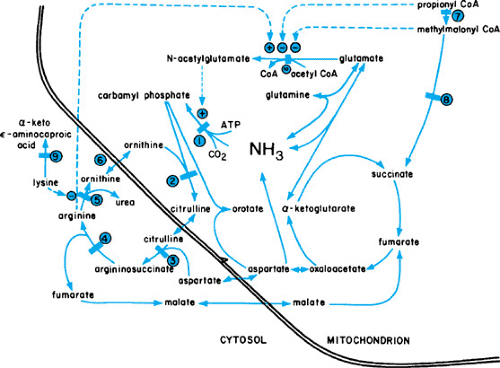 FIGURE 367.1. Urea cycle. Major metabolic pathways for the use of ammonia. Solid bars indicate sites of primary enzyme defects (or suspected sites of primary enzyme defects) in various inherited metabolic disorders associated with hyperammonemia. 1, carbamyl phosphate synthetase I; 2, ornithine transcarbamylase; 3, argininosuccinate synthetase; 4, argininosuccinate lyase; 5, arginase; 6, mitochondrial ornithine transport 7, propionyl coenzyme A (CoA) carboxylase; 8, methylmalonyl CoA mutase; 9, 4-L-lysine dehydrogenase; 10, N-acetylglutamate synthetase. Dotted lines indicate sites of pathway activation (+) or inhibition (-). ATP, adenosine triphosphate. (Reprinted with permission from Flannery DB, Hsia YE, Wolf B. Current status of hyperammonemic syndromes. Hepatology 1982;2:495.) |
Hyperammonemia may be reduced through the use of peritoneal dialysis, hemodialysis, or venovenous hemofiltration. Efforts should be made to induce ammonia excretion through alternative pathways. Therapy should be tailored to individual disorders. In addition to the administration of low-protein diets, sodium benzoate often is given to allow the excretion of ammonia in the form of hippurate. Arginine or citrulline may be given to enhance the excretion of nitrogen. Sodium phenylacetate or phenylbutyrate may be given in argininosuccinase deficiency; glutamine may be conjugated with phenylacetate, resulting in the renal excretion of phenylacetylglutamine and the accompanying excretion of nitrogen. These steps have allowed the successful management of many episodes of hyperammonemia, with longer survival of affected patients. However, catastrophic episodes still may occur despite adequate therapy. Efforts at identifying carriers and prenatal diagnosis are critical. Specific prenatal diagnosis of ornithine transcarbamylase deficiency has been described. Early, prospective treatment of at-risk infants results in more favorable outcomes. Liver transplantation may be curative if performed before irreversible neurologic injury.
Such organic acidemias as propionic acidemia, isovaleric acidemia, methylmalonic acidemia, glutaric acidemia, and multiple carboxylase deficiencies also may manifest with neonatal hyperammonemia. Typically, serum ammonia levels are elevated to a degree less than that observed in urea cycle defects, and usually acidosis and ketosis are evident. Evaluation includes a careful analysis of the pattern of urinary organic acid excretion. Therapy focuses on correcting specific defects, including acidosis and removal of ammonia through the use of dialysis. Subsequent medical or dietary therapy (or a combination) is tailored to individual disorders; hepatic transplantation
has been performed for selected disorders, however neurologic outcome may not be altered by this therapy.
has been performed for selected disorders, however neurologic outcome may not be altered by this therapy.
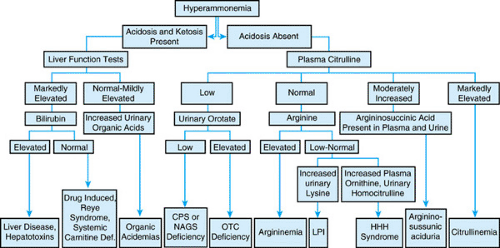 FIGURE 367.2. Differential diagnosis of hyperammonemia. CPS, carbamyl phosphate synthetase; Def., deficiency; HHH, hyperornithinemia-hyperammonemia-homocitrullinemia; LPI, lysinuric protein intolerance; NAGS, N-acetyl glutamate synthetase; OTC, ornithine transcarbamylase. (Reprinted with permission from Batshaw ML. Hyperammonemia. Curr Probl Pediatr 1984;14:1.) |
The liver is a major target organ of inherited disorders of oxidative phosphorylation. Liver disease of varying severity may occur at different ages, in association with deficiencies of the respiratory chain complexes. Neonatal liver failure has been described with deficiency of complexes III and IV. Mitochondrial DNA depletion, with a general reduction in activity of respiratory chain complexes encoded by the mitochondrial genome (complexes I, III, and IV), also has been described. These infants present within the first months of life with hypoglycemia, coagulopathy, hyperbilirubinemia, and hyperammonemia. A significant lactic acidemia and elevated lactate-to-pyruvate ratio is present. Liver biopsy specimens show microvesicular and macrovesicular steatosis. Death from liver failure is common. Liver transplantation should not be done in the presence of progressive neuromuscular disease, which occurs frequently in these patients and is not reversed through this treatment modality.
Transient hyperammonemia of infancy is a disorder found primarily in premature infants, often in conjunction with respiratory distress. Typically, ammonia levels rise rapidly during the first days of life, producing central nervous system (CNS) depression and coma in the second and third days of life. Family history is negative, unlike that often found for infants with urea cycle defects or organic acidemias. The cause of this disorder is unknown, although diminished hepatic blood flow in the neonatal period has been postulated. Therapy is aimed at reducing serum ammonia levels, and dialysis has been used. Prognosis appears to be good for patients whose disease is treated aggressively and who survive the neonatal period.
Reye Syndrome
Reye syndrome became a topic of widespread interest in the mid-1970s, when a dramatic increase in incidence was observed. Although the ensuing years have brought a better understanding of the epidemiology and therapy of Reye syndrome, its cause remains unclear. Clearer, however, is that most cases presenting as “Reye syndrome” are due to defects in mitochondrial metabolism. Despite its declining incidence, therefore, the disease continues to attract interest as a paradigm for other mitochondrial disorders.
Clinical and Laboratory Features
Classic Reye syndrome, or acute encephalopathy and fatty degeneration of the viscera, follows a biphasic course. Within a week after recovery from a generally mild prodromal viral illness, pernicious vomiting occurs, usually without fever. Irritability and lethargy may be present initially. Most patients do not progress further; however, some may display delirium and stupor followed by progression to seizure, coma, or death due to brainstem herniation. Focal neurologic findings are absent. Mild hepatic enlargement occurs, although jaundice does not. The cerebrospinal fluid is normal. The encephalopathy may continue for 24 to 96 hours, by which time gradual improvement in neurologic function begins to occur in patients who eventually recover, or deterioration in those for whom progression is obvious.
The laboratory features of this syndrome include an elevation of serum aminotransferase levels to at least threefold above normal. Serum ammonia may be normal or elevated at presentation, as may the prothrombin time; however, serum ammonia levels above 100 mg/dL and a corrected prothrombin time of 3 seconds or longer than control levels on admission are harbingers of progression to deeper coma grades. Hypoglycemia may affect primarily infants and younger children. The differential diagnosis of children presenting with a Reye syndrome–like picture includes CNS infections, salicylate toxicity, valproate and other drug toxicity, urea cycle disorders, disorders of fatty acid oxidation, and organic acidemias.
Epidemiology
Pathology
A liver biopsy is useful in confirming the diagnosis of Reye syndrome. Grossly, the liver may be yellow or white as a result of the high triglyceride content. Microscopic findings include the characteristic panlobular microvesicular fatty infiltration of hepatocytes. Inflammation is minimal, cell necrosis is rare, and little or no cholestasis is evident. The ultrastructural changes in the mitochondria, which appear to be unique to Reye syndrome, reflect changes in mitochondrial function and include matrix expansion and loss of mitochondrial dense bodies. The number of mitochondria may decrease. Additional findings include depletion of glycogen and increased numbers of peroxisomes.
Pathogenesis
The cause of Reye syndrome and associated mitochondrial dysfunction is unknown, although viral infection is related to disease onset. Salicylates have been implicated in the pathogenesis of Reye syndrome. Although Reye syndrome does not represent acute salicylate toxicity, multiple studies have suggested a higher incidence of aspirin use in patients developing Reye syndrome. Therefore, current recommendations are to withhold aspirin use in children, especially during periods of increased varicella and influenza activity.
Therapy
The treatment of Reye syndrome involves careful monitoring for progression in the early, less severe stages and intensive supportive care addressing increased intracranial pressure and the other consequences of mitochondrial dysfunction in the more severe stages. Specific therapy is unavailable.
Prognosis
The prognosis for patients with grade I disease is excellent, with rapid recovery expected. Prognostic indices include the degree of elevation of serum ammonia and prolongation of prothrombin time on admission. Neurologic deficits may occur after recovery from more severe disease; they include deficits in measured intelligence, achievement, visuomotor integration, and concept formation.
Diseases Resembling Reye Syndrome
Many diseases, including urea cycle defects and organic acidemias, may present with episodes of hyperammonemia. Although most of these disorders present primarily in infancy, some, such as the heterozygous form of ornithine transcarbamylase deficiency, may appear later in childhood with Reye syndrome–like illnesses. Other entities that may appear in infancy or childhood include medium-chain acyl-CoA dehydrogenase (MCAD) deficiency, long-chain acyl-CoA dehydrogenase deficiency, and short-chain acyl-CoA dehydrogenase deficiency; carnitine-acylcarnitine translocase deficiency; and 3-hydroxy-3-methyl glutaric acidemia. Common features shared by these defects include acute metabolic decompensation associated with fasting, chronic involvement of fatty acid–dependent tissues (e.g., cardiac and skeletal muscle), episodes of hypoketotic hypoglycemia, and an alteration in the esterification of plasma or tissue carnitine.
Carnitine aids in the transfer of long-chain fatty acids across the inner mitochondrial membrane, where they undergo beta-oxidation. Carnitine-acylcarnitine translocase deficiency is associated with hypoglycemia; with lipid accumulation in liver, skeletal muscle, and heart; and with low serum carnitine levels. Hyperammonemia and sudden death within the neonatal period also have been reported. Replacement therapy may be useful. Prenatal diagnosis is available.
Deficiencies of enzymes involved in intramitochondrial fatty acid oxidation pathways have been described. Variously, MCAD, very long-chain acyl-CoA dehydrogenase (VLCAD), and long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiencies may manifest with episodes of nonketotic coma, usually provoked by fasting. Frequently, vomiting, hypoglycemia, and hyperammonemia are observed features. Acute hepatic failure may occur with the long-chain fatty acid oxidation defects. Liver biopsy reveals large amounts of microvesicular fat. These defects in fatty acid oxidation should be suspected in any children with a history of recurrent episodes of vomiting, lethargy, coma, and hypoglycemia. Despite hypoglycemia, ketones are not present in the urine. The presentation of MCAD deficiency occurs in early childhood; that of VLCAD deficiency occurs primarily in young infants (younger than 4 months), with occurrence of LCHAD intermediate between the two. VLCAD deficiency appears to be the more severe form and may be associated with hypotonia, hepatomegaly, cardiomyopathy, developmental delay, and death. Heterozygotes for LCHAD deficiency may present as adults with acute fatty liver of pregnancy. MCAD deficiency has been associated with sudden infant death syndrome. Although this disorder previously was thought to be relatively benign, prospective studies have identified a moderate incidence of neurologic deficits in children with the disease.
Diagnostic efforts should include a search for characteristic patterns of urine dicarboxylic acid excretion during acute episodes of illness or after fasts. A total plasma carnitine concentration of less than 30 μmol/L suggests a fatty-acid oxidation disorder. Other diagnostic measures may include measurement of acyl-CoA dehydrogenase activity in liver or in fibroblasts. Postnatal DNA screening for MCAD deficiency has been advocated. Mutations responsible for VLCAD and LCHAD also have been identified. Treatment modalities are unproven and therefore remain speculative, but they have included high-carbohydrate, low-fat diets, carnitine supplementation, and use of dietary medium-chain triglyceride oil in patients with VLCAD. Avoidance of fasting is recommended. In acutely ill children, glucose should be administered at rates sufficient to prevent fatty-acid mobilization (approximately 10 mg/kg/minute) until oral feeding can be resumed. (For further discussion of disorders of fatty acid oxidation, see Chapter 386.)
Nonalcoholic Steatohepatitis (NASH)
Fatty infiltration of the liver may be seen in a wide variety of conditions, including malnutrition/malabsorption, cystic fibrosis, Wilson disease, the use of specific drugs, hypercortisolism, Reye syndrome, abetalipoproteinemia, peroxisomal and mitochondrial disease, and alcohol abuse, among others. Excluding those patients with significant concurrent alcohol use, children with fatty infiltration of the liver are said to have nonalcoholic fatty liver disease (NAFLD). Obese patients, as well as those with type II diabetes, also are at risk of NAFLD. In fact, 10% to 20% of obese children have elevated serum aminotransferases, and approximately 20% of obese children with normal aminotransferases have ultrasonographic findings that suggest fatty infiltration of the liver. The prevalence of this problem in pediatrics is underlined by the finding that 60% of adolescents found to have elevated serum aminotransferases are overweight. In a series of 24 children in whom ultrasound examination suggested fatty infiltration (the majority of whom were
obese), all had hepatic steatosis on biopsy. The mechanisms by which fat infiltration of the liver occurs presumably vary according to underlying etiology. In general, however, steatosis implies either enhanced hepatic free fatty acid delivery or synthesis, or diminished excretion/degradation, either through diminished β-oxidation or diminished VLDL synthesis or excretion. Hyperinsulinemia favors hepatic steatosis through the stimulation of fatty acid synthesis in combination with both diminished oxidation and VLDL excretion.
obese), all had hepatic steatosis on biopsy. The mechanisms by which fat infiltration of the liver occurs presumably vary according to underlying etiology. In general, however, steatosis implies either enhanced hepatic free fatty acid delivery or synthesis, or diminished excretion/degradation, either through diminished β-oxidation or diminished VLDL synthesis or excretion. Hyperinsulinemia favors hepatic steatosis through the stimulation of fatty acid synthesis in combination with both diminished oxidation and VLDL excretion.
Nonalcoholic steatohepatitis (NASH) represents a form of NAFLD in which hepatic steatosis is noted in combination with hepatic inflammation. Patients with NASH are generally asymptomatic, although right upper quadrant pain may occur, as may symptoms associated with portal hypertension (ascites, variceal hemorrhage, etc.) in advanced cases. Hepatomegaly may be noted in as many as 25% of adult patients. Serum ALT values are generally elevated twoto threefold. Unfortunately, these findings are not specific to NASH, and do not allow it to be distinguished clinically from NAFLD. Pathologic findings range from Grade 1 (steatosis, mild inflammation in the hepatic lobule) to Grade 3 (severe steatosis, lobular and portal inflammation, ballooning degeneration of hepatocytes). Severe disease also may be associated with fibrosis, which may progress to cirrhosis. Of 24 children with biopsy-proved steatosis, 88% had inflammation on biopsy, and 75% had fibrosis. Of these, seven children had fibrosis classified as moderately severe, and one child had cirrhosis. Progression to cirrhosis may be rapid—in one reported child, it occurred within a 2-year period.
The pathophysiology of NASH, especially as distinguished from NAFLD, is uncertain. Possible inciting factors include enhanced oxidant stress and direct hepatocyte toxicity from elevated serum free fatty acid levels. The latter is supported by studies in which animals deficient in peroxisome proliferator-activated receptor alpha (PPAR-α) and peroxisomal fatty acyl-CoA oxidase (AOX) develop steatohepatitis, suggesting that endogenously produced fatty acids, usually oxidized by AOX, may act as nuclear transcription factors, important in the pathogenesis of NASH. Steatohepatitis also develops in animals deficient in methionine adenosyltransferase 1A, important in the formation of S-adenosylmethionine (AdoMet). Animals depleted in AdoMet demonstrate enhanced oxidative stress and lipid peroxidation.
The mainstay of the therapy of NASH in the obese child is weight loss and exercise. In a series of 38 obese children with NAFLD inferred from ultrasound, 79% of those who lost weight demonstrated improvement or clearing of previous ultrasound findings. Similar findings have been documented in adults. Supplementation with vitamins E and C may also be beneficial, presumably because of their antioxidant properties. Betaine (a methyl-group donor) also has shown promise in preliminary studies. Hepatic transplantation may be required in severe cases.
DISORDERS OF METAL METABOLISM
Wilson Disease
Wilson disease (i.e., hepatolenticular degeneration) is an autosomal recessive disorder of copper metabolism. The clinical presentation is highly variable, but symptoms rarely are evident before age 5. Patients younger than age 20 tend to present with predominantly hepatic manifestations, such as asymptomatic hepatomegaly, an illness mimicking acute hepatitis, and with a picture similar to other forms of chronic active hepatitis. Hepatic insufficiency associated with cirrhosis may develop slowly and may manifest with signs of portal hypertension, including ascites, edema, and variceal hemorrhage. Conversely, patients may present with acute liver failure (ALF) associated with a brisk hemolytic anemia; this specific presentation is almost uniformly fatal if orthotopic liver transplantation is not performed. Older patients present with predominantly neurologic and psychiatric disturbances. Often, these disorders initially are subtle and include deterioration in school or job performance, behavioral changes, slurred speech, and tremors. If untreated, severe dysarthria and dystonia result, often leading to psychiatric hospitalization or institutionalization. Kayser-Fleischer rings (i.e., copper deposits on the inner surface of Descemet’s membrane) invariably are found if neurologic disease exists, but they may be absent in younger patients having liver disease only. Hemolysis, presumably secondary to the release of copper from the liver, may be present, as may calcified, pigmented gallstones. Other clinical features may include Fanconi syndrome, with progressive renal disease and arthritis.
Pathogenesis
The clinical manifestations of Wilson disease are thought to result from an excessive accumulation of copper in the liver, eyes, CNS, and kidneys. The reason for copper accumulation is unknown, but the most likely explanation may be the defective excretion of copper from hepatocytes into the bile, presumably secondary to an undefined lysosomal defect. A gene linked to Wilson disease has been mapped to the long arm of chromosome 13; the product of this gene is a P-type copper-transporting adenosine triphosphatase (ATPase; ATB7B; Wilson disease protein (WNDP)). Multiple mutations in this gene, resulting in the Wilson disease phenotype, have been identified; distinct mutations may be associated with distinct phenotypes. The inheritance pattern is autosomal recessive. WNDP is found primarily in the trans-Golgi apparatus of cells, where it is thought to transfer copper to copper-dependent enzymes and proteins, including ceruloplasmin. When intracellular concentrations of copper are increased, WNDP also is found in vesicles into which copper is thought to be sequestered, thus protecting the cell from toxicity. Data also indicate defective synthesis of the copper-binding protein ceruloplasmin in patients with WD. The defect appears to reside at the level of messenger RNA production. The precise cause of copper hepatotoxicity is unclear, but a postulated mechanism is the oxidation of sulfhydryl groups, which depletes stores of reduced glutathione. Copper also may inhibit a variety of enzymatic processes.
The natural history of Wilson disease begins with the asymptomatic storage of copper in hepatocytes. As saturation occurs, hepatocyte necrosis occurs, with the stored copper released into the circulation. This process may result in hemolysis and copper deposition in the eye, kidney, and CNS. Hepatic fibrosis and cirrhosis occur in conjunction with the progressive deposition of copper in other tissues.
Laboratory features of Wilson disease may include a low serum ceruloplasmin level. Serum copper may be elevated, particularly during episodes of hemolysis, and urinary copper excretion is markedly increased. The examination of urine copper excretion after a dose of D-penicillamine may be particularly valuable. Hepatic copper content is increased to more than 250 μg/g dry weight, although sampling error may be an issue with percutaneous needle biopsy specimens. Liver tissue may contain fatty infiltration, glycogen granules, and enlarged Kupffer cells. A lesion indistinguishable from that observed in autoimmune hepatitis may be observed in some patients. Advanced cases may demonstrate hepatic fibrosis and cirrhosis.
Diagnosis
The diagnosis of Wilson disease should be considered in all children with unexplained hepatic disease. A high level of clinical
suspicion must be maintained. Signs of chronic liver disease must be sought. Kayser-Fleischer rings should be sought through a careful slit-lamp examination. Neurologic signs, especially in older patients, may be found. CNS lesions may be demonstrated with CT or magnetic resonance imaging (MRI) scans. Helpful laboratory features include decreased serum ceruloplasmin levels and an elevated serum copper level; the 24-hour urinary copper excretion rate, normally less than 40 μg/24 hours, may be more than 100 μg/24 hours in Wilson disease. However, this finding may be present in other forms of chronic liver disease. To discriminate, a dose of oral D-penicillamine increases urinary copper excretion in Wilson disease to levels of 1,200 to 2,000 μg/day. Hepatic biopsy, when assessed for histologic change and hepatic copper content, is the single best method for the diagnosis of Wilson disease. In patients exhibiting the fulminant presentation of Wilson disease, serum alkaline phosphatase and serum aminotransferase levels often are disproportionately low, but the serum and urine copper content is elevated markedly. Genetic testing currently is unavailable.
suspicion must be maintained. Signs of chronic liver disease must be sought. Kayser-Fleischer rings should be sought through a careful slit-lamp examination. Neurologic signs, especially in older patients, may be found. CNS lesions may be demonstrated with CT or magnetic resonance imaging (MRI) scans. Helpful laboratory features include decreased serum ceruloplasmin levels and an elevated serum copper level; the 24-hour urinary copper excretion rate, normally less than 40 μg/24 hours, may be more than 100 μg/24 hours in Wilson disease. However, this finding may be present in other forms of chronic liver disease. To discriminate, a dose of oral D-penicillamine increases urinary copper excretion in Wilson disease to levels of 1,200 to 2,000 μg/day. Hepatic biopsy, when assessed for histologic change and hepatic copper content, is the single best method for the diagnosis of Wilson disease. In patients exhibiting the fulminant presentation of Wilson disease, serum alkaline phosphatase and serum aminotransferase levels often are disproportionately low, but the serum and urine copper content is elevated markedly. Genetic testing currently is unavailable.
Therapy
Untreated, Wilson disease is uniformly fatal. Adequate therapy is available in the form of triethylene tetramine dihydrochloride (Triene), a copper chelating agent. D-Penicillamine (beta, beta-dimethyl-cysteine) may be used alternatively, and is given orally in initially low doses, increasing to 1 g/day for adults and 0.50 to 0.75 g/day for younger children. Urinary copper excretion increases initially during chelating therapy, leading to the “decoppering” of affected patients. Urinary copper levels later stabilize, reflecting the maintenance of copper balance. In conjunction with this therapy, a diet low in copper must be instituted; foods with a high copper content, such as liver, chocolate, nuts, and shellfish, should be avoided. These restrictions should maintain the daily copper intake below 1 mg/day. Water sources should be analyzed for copper content. With the institution of therapy, usually an improvement in hepatic and neurologic function is found, and the Kayser-Fleischer rings regress. This therapeutic program must be followed for life. One study found that patients who discontinue therapy often die within 3 years. Zinc acetate, administered orally, may maintain a negative copper balance in some patients with Wilson disease, and it can be a useful form of maintenance therapy, particularly in patients unable to tolerate other chelator therapy, or in combination with chelator therapy. At present, its sole use as initial therapy in symptomatic patients cannot be recommended. Zinc appears to exert its effect primarily by decreasing the intestinal absorption of copper. Tetrathiomolybdate shows promise in the therapy of patients with preexisting neurologic disease, in whom the institution of D-penicillamine may be associated with worsening of symptoms, although data are limited at present.
For Wilson disease patients who present with ALF associated with hemolysis, intensive support is indicated; plasma perfusion, hemodialysis, and peritoneal dialysis may be efficacious in lowering the serum copper and in decreasing the copper burden. However, the hepatic injury apparently is irreversible; after stabilization, efforts should be directed toward rapid diagnosis and referral of affected patients for liver transplantation, which is curative.
Prognosis
Untreated Wilson disease is uniformly fatal; death may occur from hepatic, neurologic, renal, or hematologic complications. The prognosis for patients presenting with ALF and hemolysis is uniformly poor without liver transplantation. With proper therapy, usually the prognosis for most patients with Wilson disease is good, although individual differences in response to chelation therapy exist. Siblings of patients with Wilson disease should be screened carefully for the disease, and therapy should be instituted in asymptomatic patients.
Indian Childhood Cirrhosis
Indian childhood cirrhosis is a form of familial childhood cirrhosis that occurs primarily in Indian Hindu families, but has been described also in Central American, Middle Eastern, and West African children. Studies suggest the presence of a similar disorder in children in the United States. Typically, onset of the disease occurs in children younger than 3 years. Affected patients present with hepatomegaly, pale stools, fever, and behavioral changes. Jaundice may be evident. Typically, the hepatic disease progresses rapidly. Hepatic biopsy confirms the progression from a nonspecific early stage to one with progressive fibrosis and, subsequently, to widespread necrosis and cirrhosis. Usually, the disease is fatal within the first 5 years of life. Suggested causes include excessive dietary intake of copper, perhaps through the use of copper and brass cooking and storage vessels. A genetic defect in the North American form of this disorder, specifically, an arginine to tryptophan mutation in the protein cirhin, expressed predominately in fetal liver, has been localized to chromosome 16q22. Therapy using D-penicillamine may allow complete recovery, especially if started early in the disease course.
Neonatal Iron Storage Disease
Neonatal iron storage disease (NISD) is a poorly characterized disorder in which hepatic insufficiency develops within the first 4 to 7 days of life. Typically, affected infants are born prematurely, and an increased familial incidence appears possible. Often, a hemorrhagic diathesis is prominent. Affected patients may die within the first week of life. Pathologic examination reveals increased iron deposition in multiple organs, including the liver, pancreas, heart, and thyroid glands. The patients have no evidence of hemolytic disease. Although the respective roles of extrinsic iron, infection, and genetic predisposition remain obscure, NISD does not appear to be related genetically to hereditary hemochromatosis. Diagnosis may be made by finding hemosiderosis in minor salivary glands on biopsy of oral mucosa, or MRI demonstrating nonparenchymal iron (pancreas, heart, etc.). Effective treatment may necessitate liver transplantation. The use of an antioxidant “cocktail” including desferrioxamine, prostaglandin E1, alpha-tocopherol, N-acetylcysteine, and selenium has been reported, however no definitive evidence exists to support efficacy.
MISCELLANEOUS ERRORS OF METABOLISM AFFECTING THE LIVER
Cystic Fibrosis
Given the presence of the cystic fibrosis transmembrane regulator protein (CFTR) within the biliary epithelium, it is not surprising that the hepatobiliary system is involved in 20% to 50% of patients with CF. In most cases, this involvement is clinically insignificant but, with improved life expectancies in this disorder, more hepatobiliary complications are being reported. Scott-Jupp et al. observed among 1,100 CF patients a
progressive rise in the prevalence of clinically apparent liver disease from 0.3% in a 0to 5-year-old group to a peak of 8.7% among those between ages 16 and 20. An increased risk of hepatic disease has been associated with specific polymorphisms of the glutathione S-transferase P1 gene.
progressive rise in the prevalence of clinically apparent liver disease from 0.3% in a 0to 5-year-old group to a peak of 8.7% among those between ages 16 and 20. An increased risk of hepatic disease has been associated with specific polymorphisms of the glutathione S-transferase P1 gene.
Infants with CF may present with persistent neonatal cholestasis, often in association with meconium ileus. Infants with this syndrome may appear to have a hypoplastic extrahepatic biliary tract at operative cholangiography, and hepatoportoenterostomy occasionally has been performed because of misdiagnosis. In nonoperated patients, bile flow spontaneously resumes in time. CF should be considered in all infants with neonatal cholestasis.
The liver pathology of affected infants reflects biliary ductal obstruction, presumably by inspissated secretions. Excessive biliary mucus is associated with periportal inflammation and mild intrahepatic bile ductular hyperplasia. Older patients with CF may develop steatosis of the liver. Tender hepatomegaly may occur with right ventricular heart failure. Focal biliary cirrhosis may occur in early childhood, but it becomes more prominent in adulthood. Approximately 24% of patients who die of CF have changes consistent with focal biliary cirrhosis found during autopsy. A small percentage of patients with focal biliary cirrhosis subsequently develop multilobular biliary cirrhosis associated with jaundice, portal hypertension, and hepatic failure. Variceal hemorrhage may occur in this condition.
The management of patients with CF and significant hepatic disease focuses on the management of portal hypertension; liver cell failure is uncommon. Options include sclerotherapy, shunting, and orthotopic liver transplantation. The administration of ursodeoxycholic acid (20 mg/kg/day; higher doses may be required in infants) often results in biochemical and functional improvement in the hepatic disease associated with CF. Long-term effects and prognosis remain uncertain.
The biliary complications of CF include microgallbladder in 15% to 20% of patients and gallstones in as many as 15% of patients. Gallstone formation may involve abnormal gallbladder size and motility, excessive amount and viscosity of gallbladder and biliary mucus, and diminished bile acid output, lithogenic bile, and probable cholesterol supersaturation of bile. Stenosis of the common bile duct also may occur.
Alpha1-Antitrypsin Deficiency
Alpha1-antitrypsin, a 50-kd glycoprotein synthesized in the liver, functions as a protease inhibitor to neutralize a broad spectrum of proteolytic enzymes, although its major target protease is leukocyte elastase. Alpha1-antitrypsin accounts for approximately 80% of the serum alpha1-globulin fraction. Homozygous deficiency of alpha1-antitrypsin is associated with neonatal cholestasis and with childhood and adulthood cirrhosis; an increased incidence of primary liver cancer has been proposed. Homozygousand heterozygous-deficient persons are at risk for pulmonary disease.
Alpha1-antitrypsin occurs in more than 75 variant forms, designated as Pi phenotypes, each inherited in a codominant fashion. The normal phenotype is MM; the type associated most often with hepatic disease is ZZ. Patients with ZZ phenotypes have low serum alpha1-antitrypsin levels, usually 10% to 15% of normal. A Pi null-null phenotype has been described in which no alpha1-antitrypsin is present in the serum. Adults with cirrhosis also have been described in conjunction with MZ, M Malton, and M Duarte phenotypes.
The incidence of the PiZZ (homozygous-deficient) phenotype is estimated to be 1 in 2,000 to 4,000. The clinical manifestations of the PiZZ phenotype in children vary. Approximately 10% of these patients present with clinical evidence of neonatal hepatic disease. Another 40% to 50%, although asymptomatic, have hepatic biochemical abnormalities when tested at age 3 months. Patients presenting with neonatal cholestasis may be jaundiced in the first week of life, and acholic stools and hepatomegaly may be observed. Often, jaundice clears by the fourth month of life. Approximately equal proportions of these infants continue to have abnormal liver function and, if untreated, die by age 10 of complications related to hepatic cirrhosis; persistently abnormal hepatic function with slow progression to cirrhosis; continued mild hepatic dysfunction and fibrosis, with survival into adulthood; or resolving hepatic disease with a return to normal function.
Pathologic findings in the liver may correlate with any of the previously described clinical conditions. In the neonatal period, the hepatic lesion may be indistinguishable from that usually found in neonatal hepatitis. Cirrhosis has been reported in neonates and in preterm infants. Variable degrees of bile-duct proliferation may occur early in the disease; this specific histologic feature portends a more severe course. The characteristic hepatic lesion in alpha1-antitrypsin deficiency is the presence of diastase-resistant hepatocyte inclusions that stain positively for periodic acid–Schiff. These globules, antigenically related to alpha1-antitrypsin, occur predominantly in periportal hepatocytes. These inclusions may not be visible before age 4 months, but their size increases with age. In more advanced disease, biopsy findings may reveal extensive fibrosis, and few intrahepatic bile ducts may be found.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


