Disorders of Renal Development and Anomalies of the Collecting System, Bladder, Penis, and Scrotum
David R. Roth
Edmond T. Gonzales
DISORDERS OF RENAL DEVELOPMENT
Abnormalities of Position
Simple Ectopia
Simple renal ectopia is a condition in which a kidney is located in an abnormal position but remains on its own side of the midline. Often, associated incomplete rotation of the renal unit occurs. The most common position is in the true pelvis; less common locations are the iliac fossa or the thorax. Ectopia occurs in 1 in 500 to 1,200 live births; is more common on the left side; and most often is discovered as part of an evaluation for other abnormalities, the most common of which are other urologic, musculoskeletal, cardiovascular, gastrointestinal, and otolaryngologic anomalies. Treatment, if any, should address pathologic factors (primarily ureteral obstruction or vesicoureteral reflux) but not the position of the renal unit.
Simple Malrotation
During embryogenesis, the kidney rotates from a position in which the renal pelvis faces anterior to its usual postnatal position, which is with the renal pelvis facing medially. Disorders of renal ascent often are associated with persistence of the original orientation. A better term would be incomplete rotation rather than malrotation. Unless problems associated with this abnormality of rotation exist, such as ureteropelvic obstruction, no treatment is needed.
Fusion Anomalies
In patients with fusion anomalies, the two renal units may remain in the true pelvis and be connected by a large mass of parenchyma (cake kidney) in the midline, or they may be partially ascended and connected by a thin isthmus of tissue below the inferior mesenteric artery connecting the lower poles (horseshoe kidney). In crossed fused ectopia, one renal unit crosses the midline and fuses to the normally positioned contralateral kidney; the ureters arise from the appropriate sides of the bladder, but one crosses the midline to enter the lower segment of the fused renal units. The anomaly is seen slightly more commonly in boys than in girls, and the kidneys reside on the right side twice as often as on the left. No treatment is needed, although an increased incidence of reflux and ureteral obstruction that might require surgery is associated with this entity.
Horseshoe kidney is the most common fusion anomaly, accounting for approximately 90% of these abnormalities. It often is discovered incidentally during an evaluation of associated anomalies or urinary infection, or at autopsy. The kidneys are positioned lower than normal, with their lower poles joined by an isthmus of tissue. They are incompletely rotated, and their axes are more vertical than normal, a consistent and characteristic sign on intravenous urography (Fig. 320.1). The isthmus usually crosses the midline anterior to the great vessels below the inferior mesenteric artery, which blocked further ascent of the kidneys during fetal development. The ureters may be somewhat dilated superior to the isthmus, which they cross; however, the need for surgical repair is unusual. Associated anomalies are common findings but usually are less serious than are those related to crossed renal ectopia. Urologic evaluation should include voiding cystourethrography because reflux is a common finding. Treatment decisions are based on the same criteria as those for a normal kidney: infection, pain, and deterioration of renal function. The abnormal location of the kidneys, as well as the isthmus, which crosses the spinal column, increases the possibility of renal injury occurring in these patients. Thus, parental counseling concerning the risks of contact sports is appropriate.
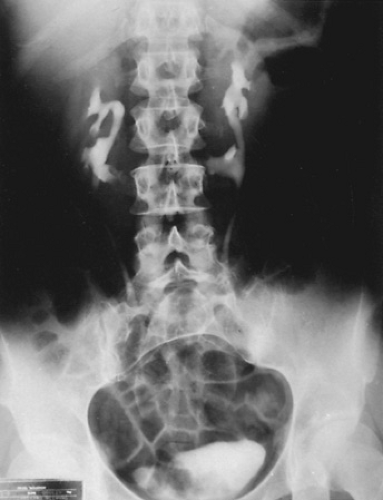 FIGURE 320.1. Intravenous pyelography of a horseshoe kidney. Note the axis deviation and incomplete rotation of the kidneys. |
Anomalies of Renal Parenchyma
Agenesis
Unilateral renal agenesis is present in 1 in 450 to 1,800 live births. Its etiology is related to maldevelopment of the metanephric duct (primitive ureter) and renal blastema. Often, the ipsilateral ureter and vas deferens are absent because their embryologic origins are similar. Compensatory hypertrophy of the solitary kidney is a common finding but not specific for this lesion. Diagnosis often is made during an investigation of other anomalies (cardiovascular, gastrointestinal, musculoskeletal). No treatment is needed, but the child and parents should be cautioned that activities that put the single kidney at undue risk of injury (e.g., organized football, rugby, riding motorcycles) should be avoided.
Bilateral renal agenesis is a rare entity that is incompatible with life. A high incidence of associated anomalies and developmental abnormalities of the bladder, urethra, and ureters occurs. Pulmonary hypoplasia is a common occurrence and generally is the immediate cause of demise. The infants have typical features, described by Potter, that include increased distance between the eyes, flattening or broadening of the nose, a prominent inner canthal fold, spade-like hands, and amnion nodosum. These findings, characteristic of Potter syndrome, are the result of severe oligohydramnios, which is caused by the absence of intrauterine urine production. This syndrome is not specific for bilateral renal agenesis, however, because any condition in which markedly decreased amniotic fluid volume occurs would produce similar findings.
Renal Hypoplasia
Renal hypoplasia is an unusual condition in which the number of renal lobules is reduced, thereby producing a kidney that is small but with normal nephron differentiation. The number of calyces is decreased, and the renal weight is diminished. If the condition is bilateral, the total nephron mass is deficient, and the result is progressive renal insufficiency with its typical complications of growth arrest and developmental delay. Patients may require renal dialysis or transplantation at any time from shortly after birth until early adulthood. Unilateral hypoplasia, on the other hand, does not cause any problems or require intervention.
Other, more common causes of small kidneys must be considered before hypoplasia is diagnosed. They include atrophy secondary to reflux nephropathy, atrophic pyelonephritis, vascular ischemia, renal vein thrombosis, and dysplasia.
Renal Dysplasia
Renal dysplasia is caused by abnormal metanephric differentiation and is a histologic rather than a clinical diagnosis. Dysplastic kidneys may involve both cystic and hypoplastic changes, although both elements not always are present. Histologically, primitive glomerular and tubular elements, cartilage, smooth muscle, and cysts are seen in the parenchyma. The dysplasia may be segmental or involve the entire renal unit. Affected parts of the kidney generally do not function. The kidney may be reniform in shape, or the dysplasia may be so severe that the unit has little resemblance to a normal kidney. Urinary tract obstruction may be associated with dysplasia and contribute to its formation.
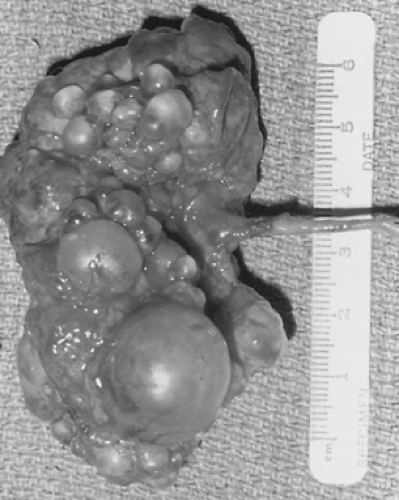 FIGURE 320.2. A gross photograph of a multicystic kidney. (Reprinted with permission from Gonzales ET Jr. Genitourinary disorders in the neonate. In: Whitaker RH, Woodard JR, eds. Paediatric urology. London: Butterworth, 1985.) |
The most common and well-known dysplastic disorder is the multicystic kidney, which consists of numerous fluid-filled cysts that do not communicate (Fig. 320.2). It is one of the two most common renal masses in the newborn (the other is ureteropelvic junction obstruction), and its diagnosis usually is suggested by ultrasound, performed either prenatally or later for evaluation of an abdominal mass. The presence of a multicystic kidney generally is confirmed by the absence of any function on a renal scan. Voiding cystourethrography should be obtained because reflux is associated in 10% of these cases. Rarely, complications occur and include urinary tract infection (UTI), rupture of renal cysts, and hypertension. The question of malignant degeneration has been raised, but the incidence of cancers is very small. Whether these lesions require nephrectomy remains controversial. Most urologists follow these children with ultrasound because, with time, almost all multicystic kidneys involute and do not seem to cause problems. Fewer physicians remove these kidneys to avoid the long-term, limited risk of development of complications and the necessity of following the children for an extended time with repeated studies.
Polycystic Kidney
Polycystic kidney disease is an inherited disorder, either autosomal recessive (infantile polycystic disease) or autosomal dominant (adult polycystic disease). The two entities are distinct and should not be confused. The recessive form is found in homozygotes, the dominant form in heterozygotes. Other organs, especially the liver, are involved; in the dominant form, cerebral aneurysms are common findings.
In the recessive form, the kidneys retain their reniform configuration but are enlarged. The parenchyma is filled with dilated renal collecting tubules that appear as small radial cysts. The collecting system (renal pelvis and ureter) is normal, as is the renal pedicle. All children with recessive polycystic kidney disease have involvement of the liver consisting of dilation and proliferation of the bile duct, with varying amounts of periportal fibrosis. Areas of uninvolved parenchyma are interspersed among these involved segments. The degree of renal and hepatic involvement appears to be inversely related, with younger children having more renal but less hepatic involvement. Children who are older when they present usually have more severe hepatic impairment, with marked periportal fibrosis. Prognosis is related to age at diagnosis; those children diagnosed at birth have the worst outcome. Those in whom the disease is found late in childhood do better, but most die before reaching adulthood, often of hepatic complications. Renal and liver transplantations have offered these patients a chance for longer survival.
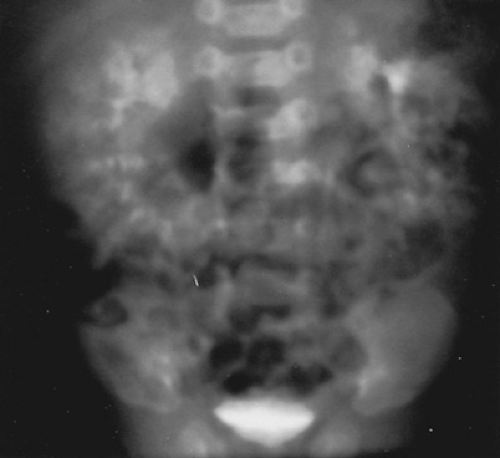 FIGURE 320.3. In this intravenous pyelogram of a child with recessive polycystic kidney disease, note the massive enlargement of the kidneys, good excretion, and linear streaking of the contrast material. (Reprinted with permission from Gonzales ET Jr. Genitourinary disorders in the neonate. In: Whitaker RH, Woodard JR, eds. Paediatric urology. London: Butterworth, 1985.) |
The recessive disease in infants can be identified prenatally by ultrasound and suspected in a family with a history of polycystic kidney disease or early childhood death from renal or unknown causes. In the neonate, the diagnosis usually is made as part of an evaluation for palpable renal masses noted on routine examination. An ultrasound shows enlarged kidneys with increased, diffuse echogenicity. Intravenous urography shows typical radial streaking of the dilated collecting tubules. Although the kidneys function, they do so poorly; without delayed films, visualization of dye in the renal pelvis, ureter, or bladder is unusual (Fig. 320.3).
The dominant form usually is noted in adults with a positive family history for renal cystic disease and is a completely different problem. It, too, is slowly progressive and ultimately results in renal insufficiency in most cases. The cysts are of various sizes and may become quite large. The kidneys may be huge and fill almost the entire abdomen. Treatment usually is limited to controlling hypertension and any infections that occur and intervening with dialysis or transplantation when necessary. A complete and thorough family history for the past several generations may identify other affected family members and assists in establishing the diagnosis. Appropriate genetic counseling should be done. Although this disorder rarely causes symptoms during childhood, small renal cysts often can be identified by renal ultrasound in affected children.
ANOMALIES OF THE COLLECTING SYSTEM
Ureteropelvic Junction Obstruction
Obstruction at the ureteropelvic junction is the most common cause of hydronephrosis in children and one of the two most common etiologies for a renal mass in neonates (the other is a multicystic kidney). The obstruction often is caused by an intrinsic fibrosis at the junction of the renal pelvis and ureter that disrupts the peristaltic wave across that region. Less common etiologies include a crossing renal vessel, kinking of the ureter, stenosis of the junction, and adhesions or extrinsic fibrosis at the ureteropelvic junction. The obstruction leads to increased intrapelvic pressure, which causes dilation of the pelvis and calyces. This obstruction predisposes to urinary stasis, infection, hematuria, pain, and gradual destruction of renal parenchyma.
The diagnosis often is suggested by prenatal ultrasound and confirmed by postnatal studies. Other signs and symptoms include UTI, pyelonephritis, abdominal or flank pain, sepsis, palpable masses, nausea, failure to thrive, or an incidental finding during the evaluation of associated congenital anomalies. Investigations should include a renal ultrasound, intravenous pyelography, or renal scan. These studies demonstrate pyelocaliectasis and late emptying of the renal pelvis (Fig. 320.4). Because intravenous pyelography often shows poor excretion, films delayed up to 24 hours may be necessary. The renal scan can estimate the relative contribution of the obstructed kidney to overall renal function, and the addition of diuresis (with furosemide) may show a prolonged washout period that suggests obstruction. Voiding cystourethrography is necessary because high-grade vesicoureteral reflux can mimic a ureteropelvic junction obstruction or cause a secondary ureteropelvic junction obstruction, as a result of the large volume of refluxed urine. In both of these cases, control of the reflux resolves the upper tract difficulties. Additionally, 10% of children with a hydronephrosis have associated reflux.
A pyeloplasty is the surgical repair of a ureteropelvic junction obstruction. Its goal is to provide a funneled and dependent pelvis leading to the ureter. Reduction of pelvic size may be necessary to facilitate renal emptying. Improvement in radiographic appearance and renal function usually occurs after relief of obstruction, although it may take years to do so.
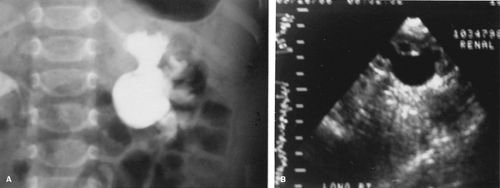 FIGURE 320.4. Ureteropelvic junction obstruction demonstrated by A: intravenous pyelography and B: renal ultrasound. |
Many patients with ureteropelvic junction obstruction are identified on prenatal ultrasound. Some of them demonstrate severe renal damage and need early surgery. The trend, though, is toward observation of neonatal hydronephrotic kidneys with good function on renal scan. Many of these kidneys improve over the course of time without surgery, demonstrating stable renal function and less hydronephrosis. Close monitoring of the renal status is necessary because occasionally a kidney deteriorates quickly and requires surgical repair. If significant hydronephrosis is present after several years, most urologists proceed to surgical correction. Neonates tolerate the surgery quite well, and, with the use of optical magnification, the procedure is overwhelmingly successful in even the youngest children. Long-term follow-up is necessary both for confirmation of an adequate postoperative anatomic result and for final assessment of renal function.
Megaureter
Ureters that are wide and dilated are called megaureters. They are divided into primary and secondary megaureters.
The primary megaureter (ureterovesical junction obstruction) is dilated, usually more distally than proximally, to the level of the ureterovesical junction, where a stenotic region, or distal inert (aperistaltic) segment, is encountered. Histologic evaluation has shown a deficiency of muscle fibers in this area that disrupts the peristaltic wave and causes functional obstruction. The usual presentations include hydronephrosis that is discovered on prenatal ultrasound or incidentally at the time of evaluation for other congenital anomalies, UTI, flank pain, or hematuria. Pyelography or ultrasound generally suggests the diagnosis. Confirmation of true obstruction usually requires that the patient undergo a diuretic renogram or, rarely, a percutaneous nephrostomy with pressure flow measurements. Voiding cystourethrography should be obtained because reflux can give the same picture on upper-tract imaging. Ureteroneocystostomy is required if obstruction is confirmed. Ureteral narrowing by tapering or tailoring may be required for an adequate repair. Generally, prognosis is good, but it depends on the extent of renal damage present at the time of surgery.
Secondary megaureters are divided into refluxing and nonrefluxing units. Those that reflux are either developmentally dysplastic or have become dilated by the volume of urine
propelled retrograde by the bladder contraction. In either case, the ureterovesical junction is incompetent and allows the reflux to occur. In most cases, surgical control of the reflux resolves the problem; however, the surgical complication rate for refluxing megaureters exceeds that for obstructed megaureters, implying that an intrinsic ureteral abnormality contributes to the ureterectasis in some cases.
propelled retrograde by the bladder contraction. In either case, the ureterovesical junction is incompetent and allows the reflux to occur. In most cases, surgical control of the reflux resolves the problem; however, the surgical complication rate for refluxing megaureters exceeds that for obstructed megaureters, implying that an intrinsic ureteral abnormality contributes to the ureterectasis in some cases.
The nonrefluxing secondary megaureter is dilated secondary to urinary obstruction at a level distal to the ureterovesical junction. The most common causes are posterior urethral valves, urethral strictures, neuropathic bladders, and dysfunctional voiding. Establishing the diagnosis may be difficult, and, because the treatments are completely different, care must be taken not to confuse the primary obstructed megaureter with the secondary form. The most reliable methods of distinguishing one from the other are diuretic renograms with a catheter in the bladder and pressure flow studies involving a percutaneous nephrostomy. In nonrefluxing secondary megaureters, control of the distal obstructive process usually solves the problem, and attention should be directed there rather than to the ureterovesical junction.
A final group of patients are those with nonobstructive, nonrefluxing megaureters. This group consists of boys with prune belly syndrome (see following discussion) and children with transient megaureters associated with endotoxins from an acute UTI. Neither condition requires treatment for the megaureter itself and, thus, an accurate diagnosis must be made to avoid unnecessary intervention.
Simple Ureterocele
A simple ureterocele is a cystic dilation of the intravesical segment of the ureter. A simple ureterocele subtends a single (nonduplicated) renal unit. These anomalies are thought to develop if incomplete dissolution of the Chwalla membrane occurs. Many simple ureteroceles are small and asymptomatic, but they can be large and obstruct the ureter or bladder neck. They usually are found when upper-tract imaging (usually a renal ultrasound) is performed to evaluate a UTI. Many are recognized on prenatal ultrasound. The radiographic findings are pathognomonic, showing a “cobra head” deformity within the bladder (Fig. 320.5). If obstruction is not present, no treatment is necessary. A transurethral incision of the ureterocele or, less commonly, ureteroneocystostomy may be required to relieve ureteral obstruction or prevent recurrent infections.
Retrocaval Ureter
The retrocaval ureter is a rare congenital anomaly in which the right ureter passes posterior and medial to the vena cava. Its etiology is related to the persistence of the subcardinal vein ventral to the developing ureter. The ureter then hooks around the future vena cava. The condition rarely is seen in children and is significant only if obstruction of the right kidney is diagnosed. Treatment consists of dividing the ureter and reanastomosing it anterior to the great vessels. Prognosis generally is excellent, with relief of the obstruction and resolution of the symptoms.
Ureteral Duplication, Ureteral Ectopia, and Ureteroceles
Complete Duplication
Ureteral duplication is the most common congenital urologic anomaly; it affects approximately 1 in 150 individuals. Duplication results when two ureteral buds arise from a single Wolffian duct. Both buds reach the developing metanephros and stimulate renal differentiation. The ureter to the lower segment is absorbed into the developing bladder earlier and, therefore, travels further along the trigone, finally resting lateral and cephalad to the upper pole ureter, which lies medial and caudal. This relationship is known as the Weigert-Meyer law. The lower-pole ureter is prone to reflux, whereas the upper-pole ureter (medial and inferior) is associated more often with obstruction from either ectopia or an ectopic ureterocele.
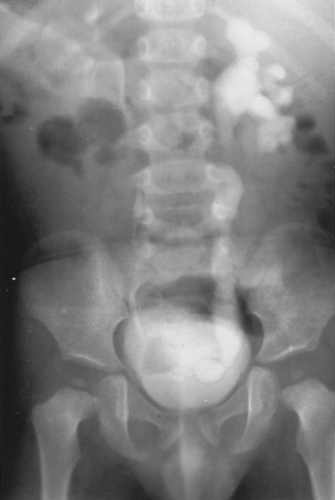 FIGURE 320.5. A simple ureterocele is shown in this intravenous pyelogram. Note the ureteral dilation and swelling of the distal ureter. |
A full spectrum of renal involvement has been observed, ranging from the child with severe bilateral lower-pole reflux and bilateral obstructing ureteroceles to the asymptomatic adult in whom duplication anomalies are discovered serendipitously. In the severe case, the diagnosis can be made after evaluation has been undertaken as a result of an abnormal prenatal ultrasound, but often it is made during the workup of a UTI or symptoms of an ectopic upper-pole ureter (see following discussion). Intravenous pyelography may show a nonfunctioning upper-pole moiety depressing the functioning lower-pole collecting system and pushing it laterally (the “drooping lily” deformity) (Fig. 320.6). A renal ultrasound shows similar findings: a hydronephrotic upper-pole cap of tissue depressing a normal lower-pole segment. The bladder may demonstrate a negative filling defect caused by nonopacification of the ureterocele. A renal scan can estimate the functional capacity of each segment, and voiding cystourethrography is required for assessment of possible lower-pole reflux.
Treatment is as varied as is the presentation. Often, when the upper pole has no function, an upper-pole partial nephroureterectomy is performed. If, on the other hand, good function exists in that segment, a transurethral incision of the ureterocele can be performed. This procedure usually relieves the obstruction, but it may cause the development of upper-pole reflux. At other times, a ureteropyelostomy connecting the upper-pole
ureter to the lower-pole renal pelvis, in conjunction with partial resection of the distal upper-pole ureter, is preferred. If lower-pole reflux is present with an upper-pole ureterocele, some surgeons reimplant the ureters at the same time the upper systems are addressed. Other surgeons delay performing any bladder surgery for several months or years, in the hope that the reflux may resolve once the ureterocele, which has been distorting the bladder, has been decompressed. When reflux to the lower pole is present without upper-pole obstruction, a common sheath ureteroneocystostomy usually is all that is required.
ureter to the lower-pole renal pelvis, in conjunction with partial resection of the distal upper-pole ureter, is preferred. If lower-pole reflux is present with an upper-pole ureterocele, some surgeons reimplant the ureters at the same time the upper systems are addressed. Other surgeons delay performing any bladder surgery for several months or years, in the hope that the reflux may resolve once the ureterocele, which has been distorting the bladder, has been decompressed. When reflux to the lower pole is present without upper-pole obstruction, a common sheath ureteroneocystostomy usually is all that is required.
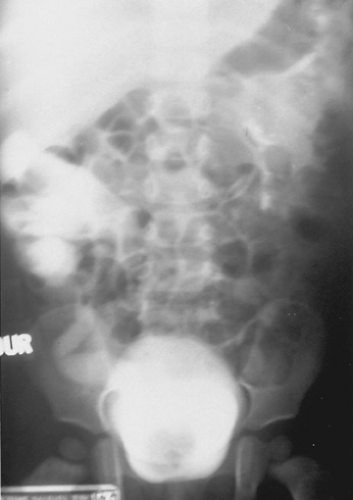 FIGURE 320.6. Intravenous pyelogram of a neonate with a right duplication anomaly. Nonvisualization of the right upper pole with the “drooping lily” deformity of the lower pole moiety is seen. (Reprinted with permission from Gonzales ET Jr. Genitourinary disorders in the neonate. In: Whitaker RH, Woodard JR, eds. Paediatric urology. London: Butterworth, 1985.) |
The prognosis depends on the degree of renal damage present at the time of intervention. However, renal function generally is adequate, and further problems seldom occur.
Incomplete Duplication
Division of the ureteral bud after it originates from the Wolffian duct causes incomplete ureteral duplication. Two ureteral buds thus promote adjacent renal differentiation but arise from a single ureteral orifice. This condition generally is asymptomatic and does not need attention (Fig. 320.7).
Ureteral Ectopia
Ureteral ectopia occurs when the ureteral orifice lies medial and inferior to its normal location. This condition can be related to either a single or duplicated drainage system. Its developmental etiology is related to the anomalous development of the terminal segment of the Wolffian duct. The origin of the ureteral bud is more cephalad than normal on the mesonephric system, precluding the usual incorporation into the trigone. In boys, the ureter can terminate along the vas deferens, seminal vesicle, prostatic urethra, or distal trigone. Because all these locations are proximal to the external sphincter, urinary continence is preserved. In girls, the analogous structures of the Wolffian duct and urogenital sinus are the bladder neck, urethra, vestibule, and Gartner duct, some of which are distal to the urinary sphincter. Ureters draining at those locations generally are associated with constant urinary leakage (Fig. 320.8), a typical presenting symptom.
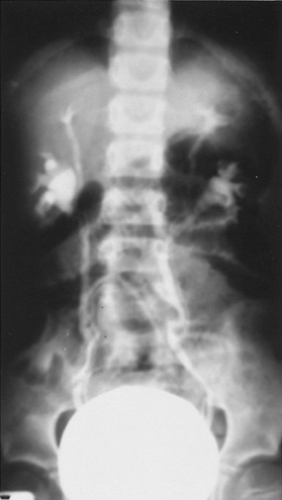 FIGURE 320.7. This voiding cystourethrogram shows bilateral partial duplication anomalies without hydronephrosis or obstruction. |
The clinical features of this disorder depend on the location of the ureteral orifice and the degree of developmental renal dysplasia. The symptoms usually are those of infection or incontinence. In girls, a history of lifelong constant wetness despite a normal voiding pattern suggests ureteral ectopia. In boys, the diagnosis of ureteral ectopia should be considered whenever a mass is found in the seminal vesicle or epididymitis or prostatitis is encountered. Radiographic studies may show either a nonfunctioning renal unit or ureteral dilation, depending on the degree of developmental abnormality. Cystoscopy can be helpful, especially if bilateral ectopia or a hemitrigone is found.
Treatment generally consists of a nephroureterectomy of the involved renal unit because renal function usually is poor. However, if kidney function is good, or in the case of bilateral ureteral ectopia, reimplantation is appropriate.
VESICOURETERAL REFLUX
Vesicoureteral reflux is the retrograde regurgitation of urine from the bladder toward the kidney. Reflux is either primary or acquired; in children, primary reflux is more prevalent. Its etiology is embryologically related to the abnormal position
of the ureteral bud on the Wolffian duct. This location causes the ureteral orifice to be lateral and cephalad on the trigone, thus foreshortening the submucosal tunnel. The tunnel normally provides the valve-like mechanism that prevents urinary reflux, and if it is deficient, reflux can occur. The degree of reflux may range from very mild (when urine enters the ureter but does not reach the kidney) to very severe (when the ureters are widely dilated and tortuous, with gross pyelocaliectasis). An objective system using well-described criteria for grading reflux is used throughout the world and is based on voiding a cystourethrography (Fig. 320.9).
of the ureteral bud on the Wolffian duct. This location causes the ureteral orifice to be lateral and cephalad on the trigone, thus foreshortening the submucosal tunnel. The tunnel normally provides the valve-like mechanism that prevents urinary reflux, and if it is deficient, reflux can occur. The degree of reflux may range from very mild (when urine enters the ureter but does not reach the kidney) to very severe (when the ureters are widely dilated and tortuous, with gross pyelocaliectasis). An objective system using well-described criteria for grading reflux is used throughout the world and is based on voiding a cystourethrography (Fig. 320.9).
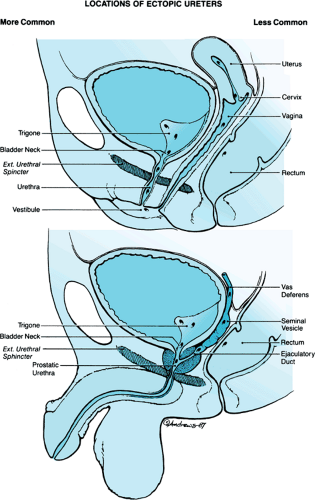 FIGURE 320.8. Possible locations of an ectopic ureter. In girls, those locations distal to the urethral sphincter allow constant wetness. In boys, however, all locations are proximal to the sphincter, so incontinence will not occur. |
The diagnosis of reflux is best made through voiding cystourethrography. Generally, this radiologic study is performed initially because it provides reproducible quantification of the reflux (which allows prognostic determination), defines anatomic anomalies at the ureteral insertion area (paraureteral diverticula, ectopia), and also visualizes the urethra, which is imperative in boys to rule out the presence of urethral obstruction. In subsequent follow-up studies, nuclear cystography may be substituted for voiding cystourethrography. The main advantage of nuclear cystography is that radiation exposure is lower compared with standard contrast cystography; however, it does not define the anatomy well, rendering grading less precise. Quantification of the reflux is important for prognosis because the lower grades of reflux tend to resolve spontaneously, whereas higher grades of reflux resolve less often and are more likely to lead to renal injury and scarring.
The basis of treatment for reflux is the premise that sterile reflux is not harmful to the kidney. Therefore, children who have reflux can be treated with a daily low-dose prophylactic antibiotic, generally nitrofurantoin or trimethoprim-sulfamethoxazole, to prevent the development of infection and to allow the kidney to grow normally. As the bladder matures, the reflux may resolve spontaneously. While the child is receiving prophylactic treatment, urinalysis and cultures should be performed every 3 to 4 months, and whenever clinically indicated, to monitor for the possible presence of infection. Cystography should be repeated at regular intervals so that, if resolution of the reflux occurs, the medications can be stopped and the child observed. Normal renal growth and development should follow; however, further infections may occur, especially during pregnancy.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


