Disorders of Purine and Pyrimidine Metabolism
Rebecca S. Wappner
PURINE AND PYRIMIDINE METABOLISM
Purine and pyrimidine nucleotides are important constituents of RNA, DNA, nucleotide sugars, and other high-energy compounds and of cofactors such as adenosine triphosphate and nicotinamide-adenine dinucleotide. Both purines and pyrimidines may be synthesized de novo from ribose-5-phosphate and carbamyl phosphate, respectively, as shown in Figs. 391.1 and 391.2. The nucleosides guanosine, adenosine, cytidine, uridine, and thymidine are formed by the addition of ribose-1-phosphate to their respective purine bases (guanine and adenine) and pyrimidine bases (cytosine, uracil, and thymine). Phosphorylation of the nucleosides results in monophosphate, diphosphate, and triphosphate nucleotides. Recycling and interconversion of the compounds occur (salvage pathways); these processes, in turn, conserve the nucleotides and nucleosides and exert a negative feedback on de novo synthesis.
The disorders of purine and pyrimidine metabolism exhibit a wide array of clinical symptoms, which include renal calculi, neurologic problems, delayed physical and mental development, self-mutilation, hemolytic anemias, and immunodeficiencies. Table 391.1 gives a summary of the findings, diagnostic testing, and treatment for the disorders.
DISORDERS OF PURINE METABOLISM
Gout
Gout is characterized by hyperuricemia, uric acid nephrolithiasis, and inflammatory arthritis. The disorder may present at any age, but most often it is seen in adults, with an increasing incidence with age. The prevalence is estimated to be 1 in 167 men and 1 in 1,000 women. Primary gout is associated with the overproduction or decreased renal excretion of uric acid. The biochemical basis of the disorder is unknown in most patients, and the disorder is considered to be a polygenic trait. Primary gout also can be seen with the overproduction of uric acid associated with increased activity of phosphoribosylpyrophosphate synthetase (PPRP-S) and deficiency of hypoxanthine guanine phosphoribosyltransferase (HGPRT), inherited disorders that are discussed in the following sections. In addition, familial juvenile gout appears to include a group of rare, inherited disorders that occur at younger ages than primary polygenic gout. Secondary gout may be seen with other conditions or disorders associated with increased production or decreased excretion of uric acid (i.e., starvation, dehydration, prolonged exercise, lactic acidosis, ketoacidosis, hypertension, renal dysfunction, myeloproliferative disorders, and glycogen storage disease type I). Secondary gout also may be seen during treatment with diuretics, low-dose salicylates, pyrazinamide, ethambutol, and niacin or during the treatment of malignant diseases. Environmental factors may play a role in the pathogenesis of gout in that excessive purine, ethanol, or carbohydrate ingestion appears to be related to increased production of uric acid.
Gouty arthritis results from monosodium urate crystal deposition in joints and surrounding tissues. The presentation usually is monoarticular and peripheral, and the most commonly affected site is the metatarsophalangeal joint of the great toe. Untreated, an acute arthritic attack resolves spontaneously within a few days to a few weeks. During acute attacks, colchicine, corticosteroids, and nonsteroidal antiinflammatory agents may be used. Chronic arthritis may lead to joint damage and deformity. Monosodium urate crystals may be noted in joint fluid. Tophi, which are monosodium urate crystal deposits, may occur over the helix of the ears and over points of insertion of tendons at the elbows, knees, and feet. Urolithiasis may occur before or after the onset of the arthritis. Calcium oxalate and urate stones are seen. Uric acid stones are yellow-orange, smooth, hard, and radiolucent, and they crush with difficulty. Renal dysfunction is thought to be related to underlying hypertension and renal vascular disease, rather than to hyperuricosuria.
Most patients with elevated uric acid levels are asymptomatic, never develop gout, and do not require long-term treatment. Symptomatic gout is more likely to develop in patients with serum uric acid levels greater than 10 mg/dL. Patients with frequent attacks of gouty arthritis, chronic gout, tophi, or uric acid nephrolithiasis may benefit from treatment
of hyperuricemia. Allopurinol, which inhibits xanthine oxidase (oxidoreductase) activity and results in reduced purine biosynthesis and reduced excretion of uric acid, is the treatment of choice for those with urate nephropathy. Probenecid and sulfinpyrazone, which increase uric acid clearance, may be used with patients with normal renal function and no uric acid renal stones. Alkalinization of the urine, which increases uric acid solubility, and increased fluid intake, which decreases the concentration of urinary uric acid, may help to prevent renal stone formation. Dietary reduction of purine-containing foods, correction of obesity, and cessation of ethanol ingestion help to lessen environmentally related causes of hyperuricemia.
of hyperuricemia. Allopurinol, which inhibits xanthine oxidase (oxidoreductase) activity and results in reduced purine biosynthesis and reduced excretion of uric acid, is the treatment of choice for those with urate nephropathy. Probenecid and sulfinpyrazone, which increase uric acid clearance, may be used with patients with normal renal function and no uric acid renal stones. Alkalinization of the urine, which increases uric acid solubility, and increased fluid intake, which decreases the concentration of urinary uric acid, may help to prevent renal stone formation. Dietary reduction of purine-containing foods, correction of obesity, and cessation of ethanol ingestion help to lessen environmentally related causes of hyperuricemia.
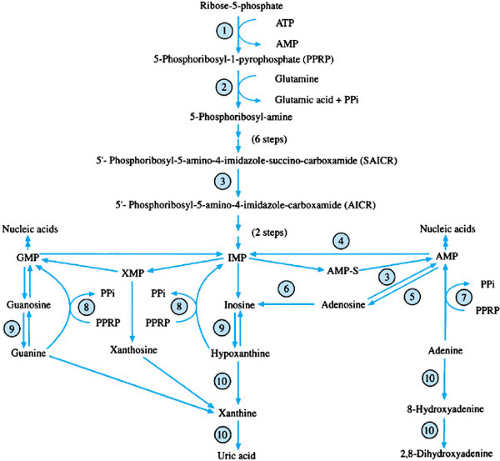 FIGURE 391.1. Purine metabolism. 1, phosphoribosylpyrophosphate synthetase; 2, amido-phosphoribosyltransferase; 3, adenylosuccinate lyase (catalyzes AMP-S to AMP); 4, adenylate deaminase; 5, purine-5′-nucleotidase; 6, adenosine deaminase; 7, adenine phosphoribosyltransferase; 8, hypoxanthine guanine phosphoribosyltransferase; 9, purine-nucleoside phosphorylase; 10, xanthine oxidoreductase. Only the ribose forms of the nucleosides and nucleotides are illustrated. The deoxyribose forms follow similar pathways. AMP, adenosine monophosphate; AMP-S, adenylosuccinic acid; ATP, adenosine triphosphate; GMP, guanosine monophosphate; IMP, inosine monophosphate; PPi, inorganic pyrophosphate; XMP, xanthosine monophosphate. |
Phosphoribosylpyrophosphate Synthetase Overactivity
PPRP-S catalyzes the transfer of the pyrophosphate group of adenosine triphosphate to ribose-5-phosphate to form PPRP. PPRP-S is induced by lowered purine nucleotide levels under normal circumstances. The resulting PPRP acts as an inducer of amidophosphoribosyl transferase, the next step in the purine biosynthetic pathway. The increased levels of purine nucleotides that result then act by means of negative feedback to inhibit purine biosynthesis. This X-linked recessive disorder is associated with reduced sensitivity of PPRP-S to nucleotide inhibition and increased specific activity of PPRP-S in vitro. The result is a continuous overproduction of purines by de novo biosynthesis. Patients have hyperuricemia and hyperuricosuria and present during the teenage or young adult years with severe gout or renal calculi. A more severe form of the disorder exists in which severe psychomotor retardation, autistic features, hypotonia, and nerve deafness also occur. Female carriers of the severe form may have deafness and gout.
Elevated PPRP levels may be detected in erythrocytes, lymphocytes, and cultured skin fibroblasts. Treatment includes allopurinol, high fluid intake, and alkalinization of the urine.
Adenylosuccinate Lyase Deficiency
Adenylosuccinate lyase (ADSL) is associated with two steps in purine metabolism. In the de novo synthetic pathway, it
catalyzes the conversion of succinyl aminoimidazole carboxamide ribotide to aminoimidazole carboxamide ribotide with release of fumarate. In the second reaction, adenylosuccinic acid is converted to adenosine monophosphate, also with release of fumarate. Deficient activity of ADSL is associated with the accumulation of both substrates for the enzyme in their nucleoside-derivative forms, succinyl aminoimidazole carboxamide riboside and succinyl adenosine. ADSL deficiency is a rare autosomal recessive disorder, associated with mutations in the ADSL gene located at chromosome 22q. Elevated succinyl purine levels are neurotoxic and result in varying degrees of psychomotor handicaps and neurologic involvement, which are usually evident by 2 years of age. More severely affected patients present with neonatal seizures. Hypotonia, autistic features, and growth retardation may occur. Computed tomography of the brain may show cerebellar hypoplasia, mainly of the vermis, and cortical and subcortical hypotrophy. Magnetic resonance imaging of the brain often shows a disturbed myelin or leukodystrophy pattern. The diagnosis is established by the finding of elevated levels of succinyl aminoimidazole carboxamide ribotide and succinyl adenosine in the plasma, urine, and cerebrospinal fluid. No effective therapy is currently available.
catalyzes the conversion of succinyl aminoimidazole carboxamide ribotide to aminoimidazole carboxamide ribotide with release of fumarate. In the second reaction, adenylosuccinic acid is converted to adenosine monophosphate, also with release of fumarate. Deficient activity of ADSL is associated with the accumulation of both substrates for the enzyme in their nucleoside-derivative forms, succinyl aminoimidazole carboxamide riboside and succinyl adenosine. ADSL deficiency is a rare autosomal recessive disorder, associated with mutations in the ADSL gene located at chromosome 22q. Elevated succinyl purine levels are neurotoxic and result in varying degrees of psychomotor handicaps and neurologic involvement, which are usually evident by 2 years of age. More severely affected patients present with neonatal seizures. Hypotonia, autistic features, and growth retardation may occur. Computed tomography of the brain may show cerebellar hypoplasia, mainly of the vermis, and cortical and subcortical hypotrophy. Magnetic resonance imaging of the brain often shows a disturbed myelin or leukodystrophy pattern. The diagnosis is established by the finding of elevated levels of succinyl aminoimidazole carboxamide ribotide and succinyl adenosine in the plasma, urine, and cerebrospinal fluid. No effective therapy is currently available.
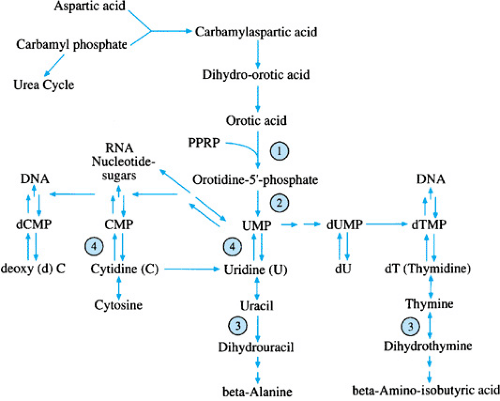 FIGURE 391.2. Pyrimidine metabolism. 1, orotic acid phosphoribosyltransferase; 2, orotidine phosphate decarboxylase; 3, dihydropyrimidine dehydrogenase; 4, pyrimidine-5′-nucleotidase, MP, monophosphate; PPRP, phosphoribosylpyrophosphate. |
Myoadenylate Deaminase Deficiency
Adenylate deaminase catalyzes the deamination of adenosine monophosphate to inosine monophosphate and is composed of multiple isoenzymes that are tissue specific. Deficient activity of muscle adenylate deaminase (myoadenylate deaminase) is an autosomal recessive disorder associated with muscle cramping and myalgia after exercise. Exercise does not lead to ammonia production, which normally would stimulate glycolysis. Muscle adenosine triphosphate and total purine content decrease to a greater extent than normally occurs with exercise. The exact metabolic abnormalities in muscle energy metabolism are not known fully. Increased creatine kinase has been noted in 60% of patients. Some patients also have hypotonia, and a few have been reported to have hyperuricemia and gout. A single mutant allele at the myoadenylate deaminase locus AMPD1, resulting in a truncated peptide, has been found in all cases evaluated to date. Myoadenylate deaminase deficiency may be shown in muscle biopsy samples by histochemical staining or measurement of specific enzymatic activity.
Secondary muscle adenylate deaminase deficiency has been reported in association with other neuromuscular disorders (i.e., hypokalemic paralysis, muscular dystrophy, motor neuron disorders, polymyositis, and other collagen-vascular diseases). Molecular genetic evaluations in these patients have shown reduced transcription of myoadenylate deaminase. Patients with muscle adenylate deaminase deficiency also appear to be at higher risk for malignant hyperthermia. In general, no specific therapy exists. Ribose administration has resulted in varying responses.
Adenosine Deaminase Deficiency
Adenosine deaminase (ADA) catalyzes the deamination of deoxyadenosine to deoxyinosine and, to a lesser extent, the deamination of adenosine to inosine. Under normal circumstances, adenosine usually is converted to adenosine monophosphate by adenylate kinase. Deficiency of ADA is associated with elevated levels of deoxyadenosine and deoxyadenosine nucleotides, especially deoxyadenosine triphosphate. These elevations lead to activation of adenosine monophosphate deaminase and cytoplasmic 5′-nucleotidase, which results in depletion of adenosine triphosphate and other adenosine nucleotides. Elevated levels of deoxyadenosine nucleotides and decreased levels of adenosine nucleotides are noted in plasma, erythrocytes, and platelets of patients. Plasma and urine levels of deoxyadenosine are markedly elevated, as are plasma levels of adenosine. The elevated levels of deoxyadenosine bind with S-adenosyl-homocysteine hydrolase and decrease its specific activity, which results in reduced methylation reactions. The elevated levels of deoxyadenosine also inhibit ribonucleoside diphosphate reductase and DNA synthesis in T-cell and B-cell precursors and are thought to be responsible for the severe combined immunodeficiency that is associated with ADA deficiency.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


