Disorders of Coagulation Factors
James F. Casella
Clifford M. Takemoto
Maria A. Pelidis
CONGENITAL ABNORMALITIES OF COAGULATION FACTORS
Abnormalities of the Factor VIII Complex
Classically, two major congenital disorders have been attributed to abnormalities of the factor VIII molecule: hemophilia A, also referred to as factor VIII deficiency, and von Willebrand disease. Hemophilia A represents a defect in factor VIII procoagulant activity in which platelet function is normal, whereas von Willebrand disease involves a defect in platelet function associated with a variable abnormality of factor VIII procoagulant activity. The abnormality of platelet function in von Willebrand disease is caused by decreased or defective von Willebrand factor (vWF), a substance necessary for platelet adhesion to damaged blood vessel walls and maintenance of a normal bleeding time.
In the past, the relation of factor VIII procoagulant activity to vWF was poorly understood. Advances in the molecular biology and protein biochemistry of factor VIII have demonstrated that circulating factor VIII is a complex of two different proteins: the factor VIII procoagulant protein (i.e., factor VIII:C) and vWF. These proteins are products of separate genes, and each has unique antigenic sites. vWF is a macromolecular structure (i.e., multimer) composed of multiple smaller subunits and appears to act as a carrier protein for the factor VIII procoagulant molecule. Therefore, factor VIII procoagulant activity is likely to be reduced when vWF is not present in sufficient quantities. The designation vWF:Ag refers to the major antigen on vWF that is recognized by heterologous antisera against the factor VIII complex; the designation vWF:RCo (i.e., ristocetin cofactor) indicates one of the activities of vWF in vitro (i.e., the ability of the molecule to support ristocetin-induced agglutination of platelets). An appreciation of these relationships is essential to understanding the clinical disease states.
Factor VIII Deficiency
Etiology and Pathogenesis
Factor VIII deficiency (i.e., hemophilia A) is a sex-linked disorder, occurring in approximately 1 in 5,000 male newborns. The disease results from a deficient or abnormal factor VIII procoagulant molecule (factor VIII:C). More than 200 discrete mutations or deletions in the factor VIII gene that result in hemophilia A have been described. However, inversions at the end of the X chromosome appear to be responsible for 35% to 45% of the cases of severe factor VIII deficiency. Spontaneous mutations are common and occur at “hot spots” in the factor VIII gene that are prone to mutations. Factor VIII levels in affected persons vary from less than 1% to approximately 25% of normal activity. Levels of vWF are normal. Female carriers of the disease are usually asymptomatic and generally have factor VIII levels between 25% and 75% of normal, with normal vWF assays. However, a carrier may have factor VIII:C activity levels higher than 100% of normal (normal range, 50% to 200%); thus, the carrier state cannot be identified in all women by use of functional assays of factor VIII:C alone. However, measurements of factor VIII:C and vWF with determinations of DNA polymorphisms among family members can detect more than 95% of the carriers of the abnormal X chromosome. Clinical severity of the disease varies with the degree of deficiency of factor VIII activity and tends to be consistent among affected male subjects in a given kindred; however, significant variations in factor VIII activity among siblings with the same mutation have been reported, a finding suggesting that the severity of the disease can be modified by other genetic factors.
Clinical and Laboratory Features
Factor VIII deficiency is characterized by a lifelong tendency toward serious and often life-threatening hemorrhage. Whereas surface bleeding and purpura can occur, deep soft tissue bleeding and hemarthrosis are the hallmarks of the disease. Patients with hemophilia can be divided into three groups based on clinical severity of the disease and the level of factor VIII activity: severe (less than 1% factor VIII activity), moderate (1% to 5% factor VIII activity), and mild (5% to 25% factor VIII activity). Patients with severe hemophilia are subject to spontaneous bleeding into joints or soft tissue sites. Those with moderate hemophilia classically develop severe bleeding only after trauma, but patients with mild hemophilia may be symptomatic only after surgery or major trauma. Life-threatening bleeding can occur in all groups. Patients with severe hemophilia may not bleed excessively immediately after small lacerations or venipunctures because of lack of impairment of platelet function; however, delayed bleeding at such sites is common, particularly if sutures have been placed.
The symptoms tend to vary with age. Approximately 50% of patients with hemophilia escape detection in the neonatal period, even if circumcisions are performed. Serious hemorrhages, including intracranial, are not uncommon in the neonatal period. Mucous membrane bleeding in the mouth and bruises, particularly palpable subcutaneous hematomas, are much more common in infancy than later life. The frequency of hemarthrosis tends to increase as the child becomes ambulatory. The age of first bleeding varies considerably. In one study, approximately 10% of patients with hemophilia suffered at least one joint hemorrhage by 11 months of age (none before 30 days of age), 27% by 18 months of age, and 33% by 30 months of age.
Although bleeding may occur at virtually any anatomic site, the most common serious bleeding encountered in hemophilia is hemarthrosis, with knees, elbows, and ankles representing the most commonly affected joints; shoulders, wrists, and hips are less frequently involved. The onset of hemarthrosis is often marked by development of pain without other objective findings, followed by acute swelling, warmth, and tenderness of the joint, sometimes accompanied by erythema or discoloration. Bleeding into soft tissues and bursae around the joint may occur. Repeated bleeding into the same joint results in synovial damage and hypertrophy, and produces secondary cartilaginous and bony abnormalities. The development of muscular atrophy and contraction of ligamentous structures around such target joints is common. The combination of soft tissue, bony, and cartilaginous abnormalities results in an anatomically abnormal joint that is more susceptible to successive bleeding episodes. Disruption of the epiphyseal structures may result in growth abnormalities. The development of bony cysts represents a late complication of hemarthrosis. Rarely, erosive pseudotumors of bone may be seen.
Central nervous system bleeding is one of the most feared complications of hemophilia and is usually the result of trauma. Symptoms may be minimal immediately after the traumatic event, and the seriousness of the bleeding may not become evident until several days after the initial incident. Even minor episodes of head trauma may be followed by intracranial bleeding, and spontaneous intracranial hemorrhage may occur.
Hemorrhage with dental procedures can be severe; preferably, patients should seek treatment from dentists who are familiar with the management of hemophilia. Lip or tongue lacerations occur frequently in toddlers and younger children and can be quite troublesome, possibly because of the high level of fibrinolytic activity of saliva. Excessive bleeding from a torn frenulum can indicate hemophilia, as does the development of a large fleshy clot (Fig. 298.1). Other gastrointestinal bleeding can occur and is usually associated with some type of structural abnormality. Bleeding into retroperitoneal spaces occurs with some frequency and can sometimes be mistaken for an intraabdominal process. Hematuria is relatively common and can be persistent. Bleeding into muscles or soft tissue can occur at any site. The seriousness of these bleeding episodes is dictated usually by their anatomic location. Entrapment of nerves or blood vessels can be particularly problematic. Bleeding in
the area of the airway should be managed as a life-threatening event. Severe hemorrhage may be experienced after surgery if adequate replacement therapy is not administered.
the area of the airway should be managed as a life-threatening event. Severe hemorrhage may be experienced after surgery if adequate replacement therapy is not administered.
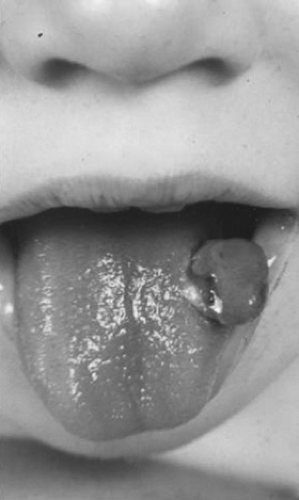 FIGURE 298.1. Granuloma formation after tongue lacceration in a patient with hemophila. |
Diagnosis of hemophilia A requires demonstration of low factor VIII:C activity in the presence of a normal vWF assay. The activated partial thromboplastin time (aPTT) usually is prolonged, and the prothrombin time (PT) is normal; however, in some mild forms of factor VIII deficiency, the aPTT result may be normal. Test results of platelet function usually are normal, although abnormal template bleeding times have been observed in some patients with hemophilia. A family history may reveal a sex-linked pattern of inheritance; however, the family history may be negative because of a predominance of female family members in successive generations or the high rate of spontaneous mutations.
Treatment, Prevention, and General Care
Prevention of bleeding should be a major goal of treatment, with care taken to avoid overprotecting the patient. Infants should be provided with padded cribs and playpens. The beneficial effects of regular exercise in strengthening muscles and protecting joints from injury should be stressed. Most practitioners recommend against contact sports, but nontraumatic sports such as swimming should be encouraged. How restrictive recommendations about sports should be is a subject of considerable debate. Blanket recommendations are often of little use, and family and patient lifestyle preferences and acceptance of risk and should be taken into account in these decisions. Platelet-inhibitory substances such as aspirin and nonsteroidal antiinflammatory drugs should be avoided. Immunizations should be administered after replacement with factor VIII or should be given intradermally rather than intramuscularly to avoid hemorrhagic complications. Vaccination against hepatitis B should be given in infancy as part of the current routine immunizations; in older children who were not vaccinated, hepatitis B immunization should be administered as soon as possible. In addition, hepatitis A vaccination is recommended for all patients with hemophilia. Prophylactic dental treatment should be encouraged. Invasive procedures such as lumbar puncture should be performed only under coverage with factor VIII.
Replacement therapy with factor VIII remains the most important part of the care of the patient with hemophilia. Home therapy has gained widespread acceptance and offers the opportunity for earlier treatment of bleeding episodes and increased autonomy for the patient. Such programs require close physician supervision. Prophylactic therapy has gained wider acceptance and should be offered to all patients with hemophilia. The goal of this therapy is to provide replacement therapy frequently enough to maintain a trough level of factor VIII greater than 1% at all times (as discussed in a following section) and to reduce long-term joint morbidity. Although expensive, this therapy can result in a significant improvement in quality of life.
Bloodborne infections such as hepatitis and acquired immunodeficiency syndrome have been major complications of therapy in hemophilia. Most patients with hemophilia who were exposed to factor VIII replacement between 1979 and 1984 are seropositive for human immunodeficiency virus (HIV). After introduction of screening procedures in 1985, the rate of HIV seroconversion in previously uninfected patients decreased dramatically. Improved methods of viral inactivation further enhanced the safety of plasma-derived factor concentrates; since 1987, there have been no documented cases of HIV transmission with these products. Recombinant factor products have been available since 1992, thus providing an extra margin of safety.
Dosage and Schedule of Factor VIII Replacement
The level and duration of replacement with factor VIII depend on the severity of bleeding. Replacement. doses can be calculated using the rule that 1 U of factor VIII/kg of body weight increases circulating factor VIII levels by 2%. For minor soft tissue bleeding, replacement to 20% of normal levels is often sufficient. For hemarthroses or more extensive soft tissue bleeding, at least 40% replacement should be achieved and may be required for several days. Levels of 70% or greater may be required for extensive dental work. Replacement of 80% to 100% is essential in the event of central nervous system bleeding or for surgical procedures. Treatment for 10 to 14 days may be required for surgical procedures or head injury.
With the first dose in a given series, factor VIII has a half-life of approximately 8 hours. Thereafter, the biologic half-life approximates 12 hours, and doses should be given at that interval to maintain a trough level one-half of the initial increment. A steady state can be achieved also by continuous infusion of factor VIII when adequate hemostasis at all times is essential (e.g., after major surgery or head injury). In these instances, a loading dose of factor VIII sufficient to raise the factor VIII level to between 50% and 100% should be given. A dosage of 3 or 4 U/kg/hour thereafter should maintain a level of approximately 50%. Considerable variation in the biologic half-life of factor VIII may be seen from patient to patient. In the event of surgery, serious hemorrhage or prolonged or continuous therapy, levels of factor VIII after infusion should be measured to determine the adequacy of replacement.
For prophylactic treatment, factor is administered on a routine schedule, usually 20 to 25 U/kg three times per week (e.g., Monday, Wednesday, and Friday, or Tuesday, Thursday, and Saturday) or every other day. Trough levels of factor VIII and the frequency of hemorrhages are useful to monitor the effectiveness of the program. Prophylactic treatment is generally considered to be optimal therapy for children with severe factor VIII or IX deficiency. The disadvantages of this approach include use of greater quantities of clotting factor concentrates, which are quite expensive. In addition, frequent venous access may be difficult for some patients, and central venous access lines such as subcutaneous ports may be required to administer the therapy. Preliminary cost-to-benefit analyses suggest that the cost of the clotting factor may be offset by the reduced arthropathy and its associated health care costs.
Preparations of Factor VIII
Recombinant factor is now considered by most practitioners to be optimal therapy for factor deficiencies and is the preferred therapy for patients with hemophilia who are naive to clotting factor replacement. Recombinant Factor VIII is purified from hamster-derived cell lines engineered to produce human Factor VIII protein. These products have virtually no risk of human bloodborne infectious agents; however, the risk of possible contamination by infectious agents still exists during production or processing. In addition, many recombinant factor VIII products are stabilized with human serum albumin, although several albumin-free products are now available. Initial concerns about the high incidence of inhibitors to factor VIII in early trials of recombinant factor VIII appear not to have been borne out, and these products have attained widespread use.
Several commercial preparations of concentrated plasma-derived factor VIII are also available. Each administration results in exposure of the recipient to tens of thousands of donors, increasing the risk of transmission of bloodborne infections. The risk of transmission of these infections has been minimized by newer techniques of preparation of factor VIII concentrates, such as detergent and wet-heat treatment. Purification and concentration of factor VIII through use of monoclonal antibodies and other techniques have resulted in production of high-purity factor VIII concentrates with low infectious risks.
Several plasma preparations contain enough factor VIII to provide at least some replacement. Fresh-frozen plasma
contains approximately 1 U/mL of factor VIII, but volume considerations and infectious risks limit its usefulness. Cryoprecipitate prepared in most blood banks contains approximately 80 to 120 U of factor VIII per bag, which can be resuspended in 10 to 20 mL of plasma or saline. Currently available methods for virus inactivation are not applicable to cryoprecipitate, and it carries a higher risk for transmission of bloodborne infections than factor VIII concentrates. For practical purposes, cryoprecipitate is of historical interest only in the treatment of hemophilia, and its use has been supplanted by factor VIII concentrates.
contains approximately 1 U/mL of factor VIII, but volume considerations and infectious risks limit its usefulness. Cryoprecipitate prepared in most blood banks contains approximately 80 to 120 U of factor VIII per bag, which can be resuspended in 10 to 20 mL of plasma or saline. Currently available methods for virus inactivation are not applicable to cryoprecipitate, and it carries a higher risk for transmission of bloodborne infections than factor VIII concentrates. For practical purposes, cryoprecipitate is of historical interest only in the treatment of hemophilia, and its use has been supplanted by factor VIII concentrates.
Adjunctive Measures for Specific Bleeding Problems
Joint Bleeding
Aspiration with irrigation of joints is not routinely done, but may be useful, especially if extreme distention of the joint capsule is encountered. If aspiration is required, coverage with factor VIII should be instituted before the procedure. Strict aseptic technique must be adhered to, because blood-filled joints may be easily infected. Prompt removal of blood from an affected joint, especially in severe bleeding, may reduce the likelihood of subsequent damage to the joint and development of target joints; however, this procedure generally requires a surgeon experienced with hemophilia. The combination of factor replacement therapy with a short course of corticosteroids (1 to 2 mg/kg/day for 3 to 5 days) may be beneficial in the event of recurrent or intractable hemorrhage or for joints in which synovial hypertrophy and synovitis have developed. Cold compresses may be applied, and brief immobilization of affected joints may provide comfort. Prolonged splinting should be avoided to prevent the development of disuse atrophy and contractures. Physical therapy should be instituted as soon as possible.
Hematuria
Although hematuria may be dramatic in hemophilia, a discrete anatomic source for the bleeding is usually not found. Administration of factor VIII is usually not effective in treating hematuria, and the administration of epsilon-aminocaproic acid is probably contraindicated because of the risk of clot formation in the ureters. Hematuria usually resolves without specific treatment. Administration of prednisone may be effective in reducing the duration and degree of spontaneous hematuria.
Dental Procedures and Mouth Bleeding
Although coverage with factor VIII remains the mainstay of therapy, epsilon-aminocaproic acid or tranexamic acid therapy may inhibit clot lysis in the mouth. Epsilon-aminocaproic acid usually is given in an oral dose of 75 to 100 mg/kg every 4 to 6 hours to a daily maximum of 24 g. Tranexamic acid is given orally at 25 mg/kg on the same schedule.
Alternative
Treatments for Mild and Moderate Hemophilia. Desmopressin acetate (DDAVP), administered intravenously or intranasally, usually increases factor VIII levels two- to fivefold in patients with detectable baseline factor VIII levels. This type of increment may be sufficient to treat minor bleeding episodes in patients with significant levels of factor VIII, but it is not useful in patients with severe hemophilia A. The usual dose for intravenous administration is 0.3 μg/kg. For intranasal adminstration, the dose is 150 μg (for patients less than 50 kg) or 300 μg (for patients greater than 50 kg). DDAVP may be administered every 12 to 24 hours. Tachyphylaxis may occur with repeated administrations. In some patients, danazol therapy has the same effect, but its use has been limited by a high frequency of side effects and unpredictable responses.
Inhibitors
Development of circulating inhibitors against factor VIII is a major therapeutic problem. High titer inhibitors are most problematic and affect 10% to 15% of patients with hemophilia. Failure to reach the expected level of factor VIII activity after infusion of factor VIII or a shortening of the biologic half-life of transfused factor VIII may be the first sign that an inhibitor is present. These inhibitors are immunoglobulin G (IgG) antibodies and often show species specificity. Their frequency does not appear to be related directly to the number of transfusions of factor VIII. Affected patients may be “low responders,” who do not increase their inhibitor level significantly with each administration of exogenous factor VIII, or “high responders,” who experience a true anamnestic response to factor VIII infusion. Low responders usually can be treated with higher doses of factor VIII. High responders may be treated at least transiently with porcine factor VIII, if their antibodies do not cross-react significantly with porcine factor VIII in vitro. Massive transfusions of human factor VIII concentrates often can overwhelm the inhibitor initially if the titer is low. Plasmapheresis may transiently lower the titer of inhibitor. Use of human and porcine factor VIII in high responders is generally restricted to instances of life-threatening emergencies or essential surgery; administration of either preparation may produce within days high titers of inhibitors that persist for long periods.
Prothrombin concentrates and activated products such as FEIBA (factor VIII inhibitor bypassing activity; Immuno-US, Inc., Rochester, MN) and Autoplex (Baxter Healthcare Corp., Glendale, CA) have had some therapeutic effect in controlled, double-blind studies and do not increase inhibitor levels. These products may be helpful in treating non–life-threatening bleeding, but the best results with these products do not approach the effectiveness of factor VIII in patients without inhibitors. Trials of NovoSeven (recombinant factor VIIa, Novo Nordisk, Princton, NJ) have demonstrated efficacy and safety in the treatment of patients with inhibitors and may become the treatment of choice for patients with high-titer inhibitors; however, this product has a short half-life requiring frequent intravenous administration as well as high cost.
Considerable progress has been reported in the use of “immune-tolerance” regimens in children with high titers of inhibitors. In many cases, tolerance to factor VIII infusions can be achieved without use of cytotoxic therapy. Continued administration of factor VIII to these patients several times each week appears to be required to prevent reappearance of the inhibitor in most cases. Considerable effort and even greater expense are involved. Whether these patients actually achieve immune tolerance instead of temporary suppression or absorption of their inhibitor is not clear. At this time, this therapy cannot be recommended for all patients with inhibitors but should be considered for patients with recent development of inhibitors, recurrent life-threatening hemorrhage, or excessive morbidity caused by an inhibitor. Treatment of inhibitors promises to continue to be one of the most vexing problems of hemophilia.
von Willebrand Disease
The term von Willebrand disease encompasses a heterogeneous group of disorders resulting from either deficiency or dysfunction of vWF. The two major functions of vWF are (a) to mediate platelet adhesion at sites of tissue injury and (b) to act as a carrier protein for factor VIII in the plasma. Abnormalities of vWF result in decreased platelet adhesiveness, impairment of agglutination of platelets in the presence of ristocetin, and prolongation of the bleeding time. Deficiency or dysfunction of vWF also results in variably decreased levels of factor VIII procoagulant activity that contribute to the coagulation disturbance. Von Willebrand disease has been classified into three major forms (Table 298.1). Mild quantitative deficiencies of vWF are referred to as type 1 or classic von Willebrand disease, which is by far the most common form, accounting for 80% to 85% of people with von Willebrand disease. Qualitative abnormalities are classified as type 2, of which four subtypes are identified (2A, 2B, 2M and 2N). Severe quantitative
deficiencies of vWF are classified as type 3. Most patients with von Willebrand disease have a mild to moderate bleeding tendency, usually involving mucocutaneous surfaces. Epistaxis, increased bruisability, and hemorrhage after dental extraction are common manifestations. Melena and menorrhagia may occur. Excessive bleeding after trauma or surgery can develop. Hemarthroses are unusual, except in type 3 disease or after significant trauma. Studies suggest that 0.8% to 1.6% of the general population shows biochemical abnormalities consistent with von Willebrand disease, thus making von Willebrand disease the most common of the inherited coagulation disorders. However, many persons with these biochemical abnormalities report no bleeding symptoms.
deficiencies of vWF are classified as type 3. Most patients with von Willebrand disease have a mild to moderate bleeding tendency, usually involving mucocutaneous surfaces. Epistaxis, increased bruisability, and hemorrhage after dental extraction are common manifestations. Melena and menorrhagia may occur. Excessive bleeding after trauma or surgery can develop. Hemarthroses are unusual, except in type 3 disease or after significant trauma. Studies suggest that 0.8% to 1.6% of the general population shows biochemical abnormalities consistent with von Willebrand disease, thus making von Willebrand disease the most common of the inherited coagulation disorders. However, many persons with these biochemical abnormalities report no bleeding symptoms.
TABLE 298.1. CLASSIFICATION OF VON WILLEBRAND DISEASE | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


