Disorders of Carbohydrate Metabolism
Rebecca S. Wappner
CARBOHYDRATE METABOLISM
Dietary carbohydrates include polymeric starch from plant sources, glycogen from animal sources, disaccharides in the form of lactose from milk sources and sucrose from fruit and vegetable sources, and, to a lesser extent, such monosaccharides as glucose, galactose, and fructose. The polymers and disaccharides are hydrolyzed to monosaccharides by enzymes present in the brush border of intestinal villi. The free monosaccharides—glucose, galactose, and fructose—are absorbed and transported to the liver, where they are used rapidly. Carbohydrate metabolism in the liver is concerned primarily with maintenance of blood glucose concentration. Glucose may be formed by the metabolism of dietary carbohydrates, from degradation of glycogen, or by gluconeogenesis from amino acids, glycerol, and lactate. Glucose-6-phosphate occupies a central position in the pathways in that all sources for free glucose first must be converted to glucose-6-phosphate except for that resulting from the action of debrancher enzyme on glycogen. In addition to being used for free glucose, glucose-6-phosphate also may be used for glycogen synthesis; for glycolysis and the production of lactate and CO2 by the Embden-Meyerhof pathway; for the production of the reduced form of nicotinamide adenine dinucleotide and CO2 by entering the
pentose cycle; and for glucuronate formation. The major disorders of carbohydrate metabolism involve the intermediary metabolism of glycogen, galactose, and fructose.
pentose cycle; and for glucuronate formation. The major disorders of carbohydrate metabolism involve the intermediary metabolism of glycogen, galactose, and fructose.
DISORDERS OF GLYCOGEN SYNTHESIS AND DEGRADATION
Glycogen Metabolism
Glycogen, the principal storage form of carbohydrate in humans, is found primarily in liver and muscle. It is a high-molecular-weight, highly branched, spherical structure composed of up to 60,000 glucosyl residues attached to a single glycogenin protein molecule. The glucosyl residues are joined in alpha-1,4-linkage to form linear chains. Branch points occur at intervals of four to ten residues where the connecting glucosyl residues are attached in alpha-1,6-linkage. Ten percent of the glucosyl residues are at the nonreducing end of the molecules; 60% of them are on outer branches, making them readily accessible for glycogenolysis. The usual concentration of glycogen in liver is less than 5 g per 100 g of wet weight. Additional information about glycogen metabolism is presented in Box 387.1.
BOX 387.1. Glycogen Metabolism
Glycogen is synthesized from glucose-1-phosphate by the actions of three enzymes (Fig. 387.1). Uridine diphosphate (UDP)-glucose pyrophosphorylase converts glucose-1-phosphate to UDP-glucose. Glycogen synthetase catalyzes the transfer of glucosyl residues from the UDP-glucose to a glycogenin protein molecule to form a glycogen primer. Glycogen synthetase also catalyzes the attachment of additional glucosyl residues in alpha-1,4-linkage to lengthen the chains. And, the brancher enzyme, amylo-1,4-1,6-transglucosylase, creates the branch points by attaching glucosyl residues in alpha-1,6-linkage to the linear chains.
Glycogen degradation involves sequential removal of nonreducing terminal glucosyl residues by a phosphorylase system and the debrancher enzyme. Phosphorylase exists in both inactive (b) and active (a) forms. Conversion of the phosphorylase to the active form requires the presence of adenosine triphosphate (ATP) and active phosphorylase kinase. Phosphorylase kinase also exists in inactive (b) and active (a) forms and is activated in the presence of ATP by a protein kinase generated in response to increased cyclic adenosine monophosphate (cAMP) formation from hormonal and chemical influences on the hepatic parenchymal cell plasma membrane. The activated phosphorylase a cleaves the alpha-1,4-linkages of the outer chains to within four glucosyl residues of the branch points and liberates glucose-1-phosphate. Then debrancher enzyme is needed for further degradation to proceed. Debrancher functions both as a transferase, which moves the three outermost glucosyl residues to another linear chain, and as an amylo-1,6-glucosidase, which then removes the final glucosyl residue in branched alpha-1,6-linkage with liberation of free glucose. In this manner, approximately 10% free glucose and 90% glucose-1-phosphate is released. The glucose-1-phosphate is converted to glucose-6-phosphate by phosphoglucomutase. Then, glucose-6-phosphate is converted to free glucose and inorganic phosphate by the glucose-6-phosphatase system.
The active site of microsomal glucose-6-phosphatase is located within the lumen of the endoplasmic reticulum. Substrates and products of its activity thus must cross the endoplasmic reticulum membrane. Five different proteins are now recognized to be necessary for normal hepatic glucose-6-phosphatase activity. The catalytic subunit of glucose-6-phosphatase hydrolyzes glucose-6-phosphate. At least four microsomal transport proteins also are involved. Microsomal transport protein T1 transports glucose-6-phosphate, the substrate for the reaction, into the lumen of the endoplasmic reticulum. Two microsomal transport proteins transport inorganic phosphate, pyrophosphate, and carbamyl phosphate across the microsomal membrane and out of the lumen where they would inhibit glucose-6-phosphatase activity. And, microsomal transport protein GLUT2, one of a family of proteins that facilitate transport of glucose across plasma membranes, transports free glucose from the microsomal lumen to the cytosol.
The synthesis and degradation of hepatic glycogen are regulated primarily by cAMP, glucose, and glycogen. An increased demand for blood glucose results in increased activation of phosphorylase and suppression of glycogen synthetase activity. Hepatic glycogen functions as a reserve for blood glucose. In contrast, muscle glycogen functions mainly as a fuel reserve for ATP generation, which is needed during exercise and usually does not contribute to blood glucose levels. Under anaerobic conditions, muscle glycogen can be degraded also to lactate by glycolysis.
Classification of Glycogen Storage Disorders
The disorders associated with abnormal synthesis and degradation of glycogen vary widely in their clinical spectrum. Overall they occur in one in 20,000 to 25,000 live births. Table 387.1 lists the disorders according to their currently accepted type and specific enzymatic or other metabolic defect. The disorders may have primarily hepatic or muscle involvement.
Hepatic Glycogen Storage Disorders
The hepatic glycogen storage disorders are estimated to occur in at least 1 in 60,000 births. Often, the pathogenesis of the various disorders may be predicted by the site of their associated enzymatic defects. All cause some degree of hepatomegaly and (usually) hypoglycemia. Functional testing may help to distinguish between the disorders. The presence of fasting hypoglycemia, the response to glucagon in the fasting and fed state, the response of blood glucose to the administration of such other carbohydrates as galactose, and the type of glucose response noted with a glucose tolerance test may be used to help differentiate between the disorders. All fasting and tolerance testing should be done with caution and close observation of the patient. For the severe disorders, the presence of lactic acidosis and hypoglycemia may be considered a relative contraindication to proceeding with fasting, glucagon stimulation,
and other tests in the classic fashion. Liver and muscle biopsies may be performed to assess the total content of glycogen and the type of glycogen structure present and to document deficient activity of specific enzymes. Open liver biopsies and muscle biopsies, rather than punch biopsies, are preferred because of the sample size needed for these analyses. Muscle biopsy should be performed simultaneously with open liver biopsy, regardless of the suspected clinical type of glycogen storage. Most important is that an experienced laboratory be contacted before obtaining the samples to ensure appropriate handling. Because many of the disorders may be documented by enzymatic analysis or by molecular genetic techniques in leukocytes, erythrocytes, or cultured skin fibroblasts, these less invasive procedures should be performed first, if possible.
and other tests in the classic fashion. Liver and muscle biopsies may be performed to assess the total content of glycogen and the type of glycogen structure present and to document deficient activity of specific enzymes. Open liver biopsies and muscle biopsies, rather than punch biopsies, are preferred because of the sample size needed for these analyses. Muscle biopsy should be performed simultaneously with open liver biopsy, regardless of the suspected clinical type of glycogen storage. Most important is that an experienced laboratory be contacted before obtaining the samples to ensure appropriate handling. Because many of the disorders may be documented by enzymatic analysis or by molecular genetic techniques in leukocytes, erythrocytes, or cultured skin fibroblasts, these less invasive procedures should be performed first, if possible.
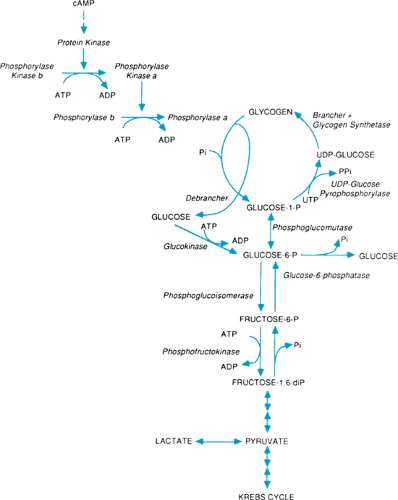 FIGURE 387.1. Glycogen synthesis and degradation. ADP, adenosine diphosphate; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; diP, diphosphatase; P, phosphatase; Pi, inorganic phosphate; PPi, inorganic pyrophosphate; UDP, uridine diphosphate; UTP, uridine triphosphate. |
All the disorders are inherited as autosomal recessive traits except for three forms of type IX glycogen storage disease (GSD), which are inherited as X-linked traits. Carrier detection and prenatal diagnosis vary, depending on the tissue distribution of the enzyme involved and whether molecular genetic testing is available for the specific disorder.
Type Ia (von Gierke Disease)
Type Ia GSD, also known as hepatorenal GSD, is associated with deficient activity of glucose-6-phosphatase in the liver, kidney, and intestine. Patients have a defective catalytic subunit of the enzyme. Affected individuals experience marked hepatomegaly, lactic acidosis, and hypoglycemia. The disorder is diagnosed most often between ages 3 and 4 months but may be recognized in the neonatal period. Milder forms may present at later ages with hepatomegaly and short stature. Other clinical features include a doll-like appearance, decreased muscle mass, renal enlargement, vomiting, diarrhea, and failure of maturation in puberty. Many affected infants are obese as a result of demanding frequent feedings, including nocturnal feedings beyond the time this behavior usually disappears (Fig. 387.2).
Fasting hypoglycemia may be profound and often results in no clinical symptoms in untreated patients. This tolerance of hypoglycemia is thought to be the result of the ability of these patients to use alternative substrates for glucose in the brain. Glucose-6-phosphatase is essential for the normal release of hepatic free-glucose, whether it is the product of glycogenolysis or gluconeogenesis. Dietary carbohydrates other than glucose also cannot be converted to glucose because the conversion involves glucose-6-phosphatase as the final step. The fasting hypoglycemia results in activation of phosphorylase and hepatic glycogenolysis, which leads to the formation of glucose-6-phosphate. The glucose-6-phosphate then is metabolized to lactate by glycolysis. Gluconeogenesis also is stimulated, and a recycling between lactate and glycogen occurs, resulting in a net increase in lactate. Stimulation of the glycolytic and gluconeogenic pathways also results in elevated triglyceride, cholesterol, very low-density lipoprotein, free fatty acid, and uric acid levels. Xanthomas, lipemia retinalis, gout, and uric acid nephropathy may occur. Biotinidase activity, frequently measured as part of newborn screening panels, is elevated in Type Ia, but not Type Ib GSD.
Liver transaminases are normal or elevated only slightly. Within the liver parenchyma, adenomatous nodules may develop, which before current dietary therapy occasionally were reported to transform into hepatic carcinoma. Abnormal bleeding tendencies that occur are thought to result from decreased platelet adhesiveness associated with the hypoglycemia. Hypercalcuria and, occasionally, renal tubular dysfunction may be present during childhood. Frequently, adults develop progressive renal disease, including focal segmental glomerulosclerosis and interstitial fibrosis.
TABLE 387.1. CLASSIFICATION OF GLYCOGEN STORAGE DISORDERS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Because patients with type I GSD frequently become hypoglycemic after 2 to 3 hours of fasting, any tolerance or stimulation test should be done cautiously and with intravenous glucose at hand. Glucose tolerance testing gives a diabetic-type early response. Fasting insulin levels are low. No glycemic response to galactose occurs (1 to 2 g/kg of 20% solution, orally or intravenously). Glucagon stimulation (20 to 30 μg/kg intramuscularly or intravenously; maximum dose, 1 mg) occasionally will produce a response, which is defined as a rise in blood glucose of 50% more than baseline. Lactic acid levels will rise with fasting and with glucagon or galactose administration. Glycogen content is elevated in the liver, kidney, and intestine. Glycogen structure is normal. On liver biopsy, the hepatocytes are noted to have glycogen in the nuclei and lipid droplets of varying size in the cytoplasm. Cirrhosis is not evident. The diagnosis may be confirmed by demonstrating a recognized mutation at the glucose-6-phosphatase gene locus in peripheral leukocytes or by measurement of glucose-6-phosphatase in liver or intestinal biopsy samples. Prenatal diagnosis and carrier detection are available only in families in whom the molecular defects are known.
Treatment is directed at supplying continuous exogenous glucose. Infants are given a formula with glucose or glucose polymers as the only carbohydrate source. Older children are given similar enteral supplements and are restricted in their intake of natural sources of galactose and fructose. Frequent daily feedings every 2 to 3 hours are supplemented with nocturnal nasogastric or gastrostomy drip feeding. Uncooked cornstarch
slurries may be used with older children and adults. Clinical and laboratory improvement is remarkable with dietary therapy. Usually, hepatic adenomas regress. Long-term survival is possible. However, affected patients continue to be at risk for significant acidosis and hypoglycemia with intercurrent illnesses or if enteral feedings are interrupted. Renal transplantation has been performed for renal failure. Liver transplantation has been shown to correct attendant biochemical abnormalities and should be considered if malignant transformation of adenomas occurs.
slurries may be used with older children and adults. Clinical and laboratory improvement is remarkable with dietary therapy. Usually, hepatic adenomas regress. Long-term survival is possible. However, affected patients continue to be at risk for significant acidosis and hypoglycemia with intercurrent illnesses or if enteral feedings are interrupted. Renal transplantation has been performed for renal failure. Liver transplantation has been shown to correct attendant biochemical abnormalities and should be considered if malignant transformation of adenomas occurs.
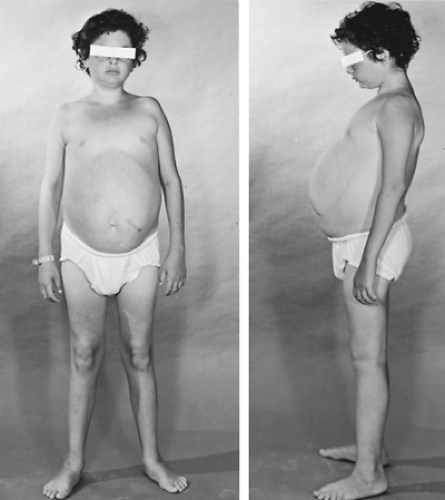 FIGURE 387.2. Glycogen storage disease Ia in a 13-year-old patient. |
Type Ib
Type Ib GSD is similar in clinical and laboratory findings to type Ia GSD. Patients also have abnormalities of neutrophil mobility and neutropenia, recurrent infections, and Crohn disease. The disorder is caused by defective transport of glucose-6-phosphate across the microsomal membrane of hepatocytes and leukocytes, owing to a defective microsomal transport protein, T1. Decreased levels of the T1 transport protein in liver biopsy samples will confirm the diagnosis. Treatment is similar to that for type Ia GSD. In addition, the neutropenia usually is responsive to recombinant human granulocyte-monocyte colony-stimulating factor or granulocyte colony-stimulating factor. Patients also will need the care of a pediatric hematologist and gastroenterologist.
Type Ic
Type Ic GSD has been reported in a few patients with deficient microsomal transport of inorganic phosphate and pyrophosphate. The clinical features are similar to that for type Ia GSD.
Type II (Pompe Disease)
Pompe disease is associated with deficient activity of lysosomal alpha-glucosidase (acid maltase) and generalized storage of glycogen. Major clinical findings include progressive myopathy, cardiomyopathy, and hepatomegaly. The disorder is discussed with the lysosomal storage disorders in Chapter 389.
Type III (Cori Disease)
Type III GSD is associated with deficient activity of the debrancher enzyme amylo-1,6-glucosidase. The disorder also is known as limit dextrinosis because glycogen of abnormal structure, similar to limit dextrin with short outer chains, is stored in liver and muscle. Most affected patients have both hepatic and muscle involvement. During early childhood, hepatic involvement predominates, and patients present in the first year of life with fasting hypoglycemia, hepatomegaly, and growth retardation. Occasionally, myopathy or cardiomyopathy occurs. No renal enlargement is noted, but renal tubular dysfunction may be seen. Mild inflammatory disease of the liver and cirrhosis may occur.
As adults, the patients often develop a progressive myopathy with abnormal electromyographs and nerve conduction velocities. Cardiomyopathy may occur. Some patients with debrancher deficiency, however, have only hepatic involvement and do not develop progressive muscle involvement.
Functional studies with glucose tolerance testing will reveal a diabetic-type early response. The response to glucagon varies but usually is normal in the fed state; no response is seen in the fasted state. Fasting lactate levels are normal but become elevated after a glucose load. Fasting ketonuria and elevated liver transaminations are seen commonly, which help to distinguish type III from type I GSD. Blood levels of cholesterol and uric acid may be elevated. Glycogen content is elevated in liver, muscle, and erythrocytes. The glycogen structure is abnormal. Liver biopsy samples have more nuclear glycogen and fewer lipid droplets than are seen with type Ia GSD. Often, fibrous septa are present. Treatment includes frequent feedings that are high in protein. Carbohydrate intake is not restricted because gluconeogenesis is intact. Galactose and fructose are converted readily to glucose. During childhood, however, nocturnal drip feedings may be required for fasting hypoglycemia. Usually, hepatic symptoms improve with age.
The diagnosis may be established by showing deficient activity of debrancher in many tissues, including leukocytes, erythrocytes, and cultured skin fibroblasts, and in liver and muscle biopsies. DNA-based diagnosis also is available. Confirmation of the risk for significant muscle involvement before clinical symptoms are present, however, can be made only by demonstrating debrancher deficiency in muscle biopsies in addition to other tissues.
Type IV (Andersen Disease)
Type IV GSD is associated with deficiency of the brancher enzyme amylo-1,4-1,6-transglucosylase. The disorder also is known as amylopectinosis because the glycogen stored has fewer than the normal number of branch points and long outer chains similar to what is seen in amylopectin. The decreased branching makes the glycogen less soluble. The cirrhosis seen in this disease is thought to result from a foreign-body reaction to the abnormal glycogen. Usually, affected infants are normal at birth but develop hepatomegaly and failure to thrive within a few months of age. Often, progressive fibrosis of the liver leads to cirrhosis, splenomegaly, and liver failure. Liver transplantation has been performed with variable outcomes. Some patients present with poor motor development, hypotonia, muscle atrophy, and absent reflexes as major clinical manifestations. Cardiac involvement may occur and can be severe. Many severely affected patients die by age 4 years. Patients with milder cases have a longer lifespan and, in some, the condition even improves with age. Usually, liver, muscle, and erythrocyte glycogen concentrations are normal, but the glycogen has an abnormal structure. The enzymatic defect may be demonstrated in leukocytes, cultured skin fibroblasts, and liver. Carrier detection and prenatal diagnosis are available.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


