Disorders of Amino Acid and Organic Acid Metabolism
Rebecca S. Wappner
DISORDERS OF PHENYLALANINE AND TYROSINE METABOLISM
Phenylalanine is an essential amino acid in that it cannot be synthesized in the body. Tyrosine is considered nonessential in normal individuals because it may be synthesized from phenylalanine. Both phenylalanine and tyrosine are present in natural foods; the amount varies with the protein content of the food source. Phenylalanine and tyrosine are precursor amino acids for such important compounds as thyroid hormone, neurotransmitters, and melanin. The metabolism of phenylalanine and tyrosine is shown in Figure 385.1.
Phenylketonuria
Phenylketonuria (PKU), perhaps the most recognized inborn error of metabolism, results from deficient activity of phenylalanine hydroxylase (PAH). It occurs in 1 in 12,000 births and is seen most frequently among persons of white and Asian backgrounds. Affected patients appear normal at birth. Untreated, they gradually develop severe psychomotor retardation, microcephaly, hyperreflexia, seizures, autistic-appearing behaviors, eczematoid-appearing rashes, and pigment dilution over the first few years of life. Intelligence quotients that are normal at birth fall to an average of approximately 60 by age 1 year and to an average of approximately 40 by age 4. Before newborn screening, most patients with PKU were diagnosed during an evaluation for delayed psychomotor development.
Untreated patients have markedly elevated blood and urine levels of phenylalanine and phenylalanine metabolites, such as phenylacetic acid and phenylpyruvic acid, which are known also as phenylketones. Levels of tyrosine are normal or low-normal. The elevated levels of phenylalanine are thought to interfere with brain growth and myelination. Increased levels of phenylacetic acid will result in a distinctive “mousy” body and urine odor. A positive blue-green color with ferric chloride urine testing indicates the presence of elevated levels of phenylpyruvic acid. Usually, enzymatic testing for PAH activity is not performed to confirm the disorder, because enzymatic activity is expressed only in liver. Mutations at the PAH genetic locus may be shown with peripheral blood leukocytes.
Newborn screening for PKU occurs throughout the United States. At birth, the infant’s blood phenylalanine level reflects that of the mother. As protein-containing feedings of breast milk or infant formula are introduced, the affected infant’s blood phenylalanine rises. Samples for newborn screening are
usually taken after 24 hours of feedings. The exact requirement, based on the age of the infant or the duration of feedings, will vary from state to state, depending upon the testing laboratory’s method and upper limits of normal (cutoff). Infants screened earlier than recommended should have repeat testing done prior to 1 week of age. Ill neonates should have testing obtained no later than 1 week of age, regardless of feeding status. Mobilization of body stores for caloric needs and intravenous feedings containing routine amino acid mixtures usually will sufficiently load the phenylalanine pathway to give a valid result. Neonates with positive screening should be referred promptly to a program or clinic that specializes in PKU care for confirmation and treatment of their disorder. The best outcomes occur in those patients in whom control of blood phenylalanine levels occurs before 2 weeks of age and in those whose blood phenylalanine levels are kept continuously within recommended treatment ranges. With treatment, all symptoms of the disorder are reversible except for loss of intellectual potential, which usually is not reversible once it has occurred. Special medical foods, low in or devoid of phenylalanine and containing tyrosine, are used. Small, measured amounts of natural foods also are given to supply the amount of phenylalanine necessary for growth. Blood phenylalanine levels are monitored frequently and dietary adjustments made to keep the phenylalanine level within the recommended control range for age. Dietary prescriptions are individualized for affected persons and their tolerance of phenylalanine. The degree of control of blood phenylalanine levels, especially during early childhood, correlates with intellectual outcome. To achieve and maintain phenylalanine control requires continuing education and support of affected patients and their families.
usually taken after 24 hours of feedings. The exact requirement, based on the age of the infant or the duration of feedings, will vary from state to state, depending upon the testing laboratory’s method and upper limits of normal (cutoff). Infants screened earlier than recommended should have repeat testing done prior to 1 week of age. Ill neonates should have testing obtained no later than 1 week of age, regardless of feeding status. Mobilization of body stores for caloric needs and intravenous feedings containing routine amino acid mixtures usually will sufficiently load the phenylalanine pathway to give a valid result. Neonates with positive screening should be referred promptly to a program or clinic that specializes in PKU care for confirmation and treatment of their disorder. The best outcomes occur in those patients in whom control of blood phenylalanine levels occurs before 2 weeks of age and in those whose blood phenylalanine levels are kept continuously within recommended treatment ranges. With treatment, all symptoms of the disorder are reversible except for loss of intellectual potential, which usually is not reversible once it has occurred. Special medical foods, low in or devoid of phenylalanine and containing tyrosine, are used. Small, measured amounts of natural foods also are given to supply the amount of phenylalanine necessary for growth. Blood phenylalanine levels are monitored frequently and dietary adjustments made to keep the phenylalanine level within the recommended control range for age. Dietary prescriptions are individualized for affected persons and their tolerance of phenylalanine. The degree of control of blood phenylalanine levels, especially during early childhood, correlates with intellectual outcome. To achieve and maintain phenylalanine control requires continuing education and support of affected patients and their families.
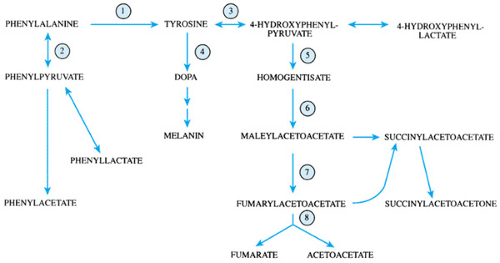 FIGURE 385.1. Metabolism of phenylalanine and tyrosine. 1, phenylalanine hydroxylase; 2, phenylalanine aminotransferase; 3, tyrosine aminotransferase; 4, tyrosine hydroxylase; 5, 4-hydroxyphenylpyruvate dioxygenase; 6, homogentisic acid oxidase; 7, maleylacetoacetate isomerase; 8, fumarylacetoacetate hydrolase. DOPA, 3,4-dihydroxyphenylalanine. |
Dietary control of blood phenylalanine levels, including the use of special medical foods, is needed indefinitely. Children and adolescents who discontinue the special diet have increasing problems with “executive” planning skills, school performance, and behavior. Adults not in dietary control have problems retaining employment, unusual personalities, panic attack–like episodes, and other neuropsychiatric problems. Actual (not just performance) intelligence may decrease. A few patients have had an acute demyelinating encephalopathy. Magnetic resonance imaging reveals “dysmyelination,” which may improve with dietary control. Other patients with PKU discontinue their special medical foods and continue to take a very low natural protein diet. These patients are at additional risk of developing multiple nutritional deficiencies, especially of cobalamin, folate, and calcium.
PKU is inherited as an autosomal recessive trait. Multiple mutations have been found at the PAH gene locus at chromosome 12q24.1. Certain mutations are more common with specific ethnic backgrounds. However, no single mutation is most common among white populations, as in cystic fibrosis. Many individuals with PKU are, in fact, compound heterozygotes for two different mutations. Carrier detection and prenatal diagnosis are available by using molecular genetic techniques in families in whom the exact mutation or polymorphisms at the PAH locus are known.
Certain mild variants of PKU allow a nearly normal dietary phenylalanine intake and do not require the use of special medical foods. Blood phenylalanine levels are elevated persistently (greater than 2 mg/dL) but are lower than the levels seen with classic PKU. Affected patients have at the PAH locus different mutations that allow for some residual activity of PAH. Importantly, girls and women with milder variants of PKU should be followed for the long term because they may have a risk of having offspring with maternal PKU syndrome (discussed in the next section) if their blood levels are elevated to more than 5 mg/dL during pregnancy.
Maternal Phenylketonuria Syndrome
The offspring of women with uncontrolled PKU during pregnancy have a significantly increased risk for cardiac malformations, microcephaly, psychomotor retardation, poor prenatal and postnatal growth, and an unusual facies, similar to that seen with fetal alcohol exposure. An increased risk for miscarriage also is present. During pregnancy, phenylalanine concentrations in the fetus are approximately 1.5 times that of the mother. Thus, if the mother’s phenylalanine levels are more than the currently recommended range of between 1 mg/dL and 3 to 5 mg/dL during pregnancy, those of the fetus will be
even higher and may reach a level that is known to be teratogenic and affect brain growth and development in the fetus. The psychomotor development of the offspring has been shown to be inversely related to the mother’s blood phenylalanine level during pregnancy. This risk occurs because of the mother’s metabolic disorder, regardless of the PAH status of the fetus. All offspring of PKU mothers will be carriers for PKU; approximately 1 in 150 offspring also will have PKU, depending on the carrier status of the father.
even higher and may reach a level that is known to be teratogenic and affect brain growth and development in the fetus. The psychomotor development of the offspring has been shown to be inversely related to the mother’s blood phenylalanine level during pregnancy. This risk occurs because of the mother’s metabolic disorder, regardless of the PAH status of the fetus. All offspring of PKU mothers will be carriers for PKU; approximately 1 in 150 offspring also will have PKU, depending on the carrier status of the father.
Improved outcomes can be seen with control of the mother’s phenylalanine level, optimally before and throughout the pregnancy. A great deal of commitment and effort is needed, however, to achieve this degree of control of phenylalanine levels during pregnancy. Women with mild forms of PKU also may be at risk (as discussed in Phenylketonuria, earlier). Women with all forms of PKU should receive counseling concerning their risks for maternal PKU syndrome, optimally before planning a pregnancy or becoming pregnant.
Tetrahydrobiopterin Synthesis and Recycling Defects
Tetrahydrobiopterin is a cofactor for PAH and for tyrosine hydroxylase and tryptophan hydroxylase. The synthesis and recycling of tetrahydrobiopterin is shown in Figure 385.2. Approximately 1% to 2% of patients with elevated phenylalanine levels will have defects in the synthesis or recycling of tetrahydrobiopterin. Deficient activities of guanosine triphosphate cyclohydrolase or of 6-pyruvoyl tetrahydropterin synthase are synthetic defects that result in tetrahydrobiopterin deficiency. Deficient activity of dihydropteridine reductase results in failure of recycling of tetrahydrobiopterin. In addition to having PKU, affected patients develop progressive neurologic deterioration during infancy as a result of decreased production of the neurotransmitters dopamine (3,4-dihydroxyphenylalanine) and 5-hydroxytryptophan. All infants with persistent hyperphenylalaninemia should be tested for biopterin defects, a procedure easily performed on filter-paper samples of blood and urine. Untreated, most affected infants die by age 1 year. Treatment with lowered phenylalanine diets will improve the elevated phenylalanine levels but will not improve the neurologic status. The use of tetrahydrobiopterin, folinic acid, and neurotransmitter replacement has resulted in varied outcomes. The disorders are inherited as autosomal recessive traits.
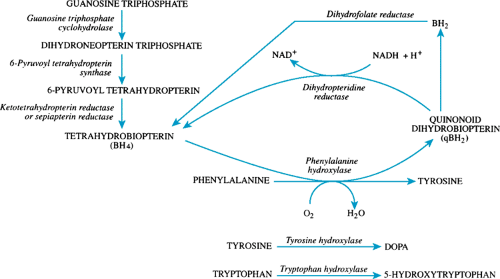 FIGURE 385.2. Biopterin synthesis and recycling. DOPA, 3,4-dihydroxyphenylalanine; NAD+, oxidized form of nicotinamide adenine dinucleotide; NADH, reduced form of nicotinamide adenine dinucleotide. |
Tyrosinemia
Elevated levels of tyrosine, or hypertyrosinemia, may occur with inherited disorders of tyrosine metabolism, severe liver dysfunction, scurvy (ascorbic acid deficiency), hyperthyroidism, or malnutrition.
Hepatorenal or Hereditary Tyrosinemia
Hepatorenal tyrosinemia, also known as hereditary tyrosinemia or tyrosinemia type I, results from deficient activity of fumarylacetoacetate hydrolase (FAH). The disorder is inherited as an autosomal recessive trait and is seen most commonly in Scandinavia and in the Saguenay-Lac St. Jean region of Quebec, where the incidence is 1 in 1,846 births. In the general population, it occurs in 1 in 100,000 to 120,000 births. Originally, the disorder was named after the markedly elevated levels of tyrosine and tyrosine metabolites found in body fluids of affected patients. Clinical manifestations seen in affected patients, however, now are thought to result from elevated levels of succinylacetone and succinylacetoacetone, which accumulate as a result of the deficient activity of FAH.
Hepatorenal tyrosinosis is a progressive multisystem disorder affecting the liver, kidneys, and peripheral nervous system. Progressive hepatic dysfunction with marked impairment of synthetic function starts in infancy. Transaminase levels may be normal or elevated only slightly. Jaundice rarely is seen early in the disease. Some patients have a rapidly progressing course and develop hepatic failure by 1 year of age. Others have acute hepatic crises that often are preceded by intercurrent illness. Markedly abnormal liver enzymes, jaundice, ascites,
and gastrointestinal bleeding commonly are seen. An unusual cabbage-like odor may be noted. Alpha-fetoprotein levels are elevated from birth and become elevated even further (100,000 to 400,000 ng/mL) with crises. Although acute hepatic crises are most likely to occur during infancy, they may happen at any age. An acute hepatic crisis is the most common type of presentation for affected patients who live in areas without newborn screening for tyrosinemia. Affected patients who survive early infancy have progressive macronodular cirrhosis. Hepatic nodules may develop and transform into hepatocellular carcinoma. Hepatocellular carcinoma occurs in approximately 18% of patients older than age 2 years; it has been reported as early as age 33 months. Liver transplantation may be needed for acute or chronic hepatic failure or for hepatocellular carcinoma.
and gastrointestinal bleeding commonly are seen. An unusual cabbage-like odor may be noted. Alpha-fetoprotein levels are elevated from birth and become elevated even further (100,000 to 400,000 ng/mL) with crises. Although acute hepatic crises are most likely to occur during infancy, they may happen at any age. An acute hepatic crisis is the most common type of presentation for affected patients who live in areas without newborn screening for tyrosinemia. Affected patients who survive early infancy have progressive macronodular cirrhosis. Hepatic nodules may develop and transform into hepatocellular carcinoma. Hepatocellular carcinoma occurs in approximately 18% of patients older than age 2 years; it has been reported as early as age 33 months. Liver transplantation may be needed for acute or chronic hepatic failure or for hepatocellular carcinoma.
Varying degrees of proximal and distal renal tubular dysfunction may occur, with associated rickets and renal tubular acidosis, glomerulosclerosis, and renal failure. Nephromegaly is noted in 80% of patients; 33% have nephrocalcinosis. A few patients have developed hypertrophic cardiomyopathies or pancreatic islet cell hyperplasia.
Elevated levels of succinylacetone are known to inhibit delta-aminolevulinic acid dehydratase activity. The resultant elevated levels of delta-aminolevulinic acid are neurotoxic and produce acute episodes of neurologic involvement similar to those seen with the porphyrias. Approximately 42% of patients develop acute episodes of neurologic crisis that most often begin as painful paresthesias. Autonomic involvement with hypertension, tachycardia, and ileus may occur. One-third of patients with neurologic crises develop a progressive paralysis that may require assisted ventilation. Peripheral nerves show axonal degeneration and secondary demyelination. Repeated neurologic crises may lead to chronic weakness. Most affected children are developmentally normal. Psychomotor regression is rare unless an acute hepatic encephalopathy develops from hepatic failure.
Dietary therapy with special medical foods devoid of or low in phenylalanine, tyrosine, and methionine along with limited amounts of natural protein from formulas or foods results in improved tyrosine levels. However, diet therapy alone will not stop the production of succinylacetone, prevent the progression of liver and kidney disease, or reduce the risk for neurologic crises or hepatocellular carcinoma. Treatment with 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), an inhibitor of 4-hydroxyphenylpyruvate dioxygenase, reduces succinylacetone production and should be started, along with dietary therapy, once the disorder is confirmed. NTBC therapy has been shown to improve hepatic and renal function and to eliminate neurologic crises. Even with NTBC therapy, affected patients still may go on to develop hepatocellular carcinoma and require liver transplantation.
Elevated levels of urinary succinylacetone and succinylacetoacetone may be demonstrated with special gas chromatography–mass spectroscopy techniques. Deficient activity of FAH may be documented in erythrocytes, peripheral blood lymphocytes, or liver biopsy samples. Newborn screening for tyrosinemia type I may be accomplished by measurement of tyrosine or succinylacetone levels in dried blood filter-paper cards. Mutations in the FAH gene locus, at chromosome 15q23-q25, have been shown in affected patients. Carrier testing may be done if the mutation or polymorphisms at the FAH locus are known. Prenatal diagnosis is available.
Oculocutaneous Tyrosinemia
Oculocutaneous tyrosinemia, also known as type II tyrosinemia or the Richner-Hanhart syndrome, is associated with deficient activity of tyrosine aminotransferase. Symptoms most often start during the first year of life; however, presentation has been reported as late as age 38. Affected patients develop a corneal dystrophy with erosions, ulcerations, opacities, and plaques. Painful, hyperkeratotic plaques develop on the palms and soles and occasionally are seen at the elbows, knees, and ankles. Approximately one-half of affected patients have psychomotor handicaps. Levels of tyrosine and tyrosine metabolites (i.e., 4-hydroxyphenylpyruvate, 4-hydroxyphenyllactate, 4-hydroxyphenylacetate, N-acetyltyrosine, and 4-tyramine) are elevated in body fluids. Treatment with a lowered tyrosine and phenylalanine diet results in clinical improvement. The disorder is inherited as an autosomal recessive trait and results from mutations at the TAT gene locus at chromosome 16q22.1-q22.3. Deficient activity of tyrosine aminotransferase may be shown in liver biopsy samples. Affected patients may be detected by newborn screening programs that include measurement of tyrosine levels.
Disorders Involving 4-Hydroxyphenylpyruvate Dioxygenase
The conversion of 4-hydroxyphenylpyruvic acid to dihydroxyphenylacetic acid (homogentisate) is catalyzed by 4-hydroxyphenylpyruvate dioxygenase (pHPPD). Defects in pHPPD have been associated with three disorders: primary deficiency of pHPPD, Hawkinsinuria, and transient neonatal tyrosinemia.
Deficient Activity of 4-Hydroxyphenylpyruvate Dioxygenase
Primary deficiency of pHPPD is a rare disorder characterized by seizures, psychomotor retardation, and episodes of acute ataxia and lethargy. Hepatic and oculocutaneous problems do not occur. Elevated levels of 4-hydroxyphenyllactate, 4-hydroxyphenylacetate, and 4-hydroxyphenylpyruvate are noted on urine organic acid profiles. Deficient activity of pHPPD may be shown in biopsy samples of liver or kidney.
Hawkinsinuria
The pHPPD reaction is complex and involves the formation of epoxy intermediates. This rare disorder is associated with the accumulation of epoxy intermediates, which may combine with glutathione to produce (2-L-cystein-S-yl-1,4-dihydroxycyclohex-5-en-1-yl)-acetic acid. This compound was named hawkinsin after the first family in whom the disorder was reported. Depletion of glutathione results in increased production of 5-oxoproline, which may be responsible for the acidosis and hemolysis noted in affected infants. Affected patients may be asymptomatic or present during infancy with failure to thrive and metabolic acidosis. One reported patient had hemolytic anemia. A chlorine-like, or “swimming pool,” odor may be noted. Plasma tyrosine is normal or elevated mildly. Most symptomatic patients respond to a lowered protein diet and supplemental ascorbic acid, a cofactor for pHPPD.
Transient Neonatal Tyrosinemia
As many as 10% of premature infants and some full-term neonates have transient immaturity of pHPPD. The relatively higher protein intake with infant formula as compared to human breast milk and a lowered intake of dietary ascorbic acid at this age also may contribute to the pathogenesis of the disorder. Many affected infants are asymptomatic, although lethargy, feeding problems, prolonged jaundice, and metabolic acidosis may occur. Elevated blood and urine levels of phenylalanine, tyrosine, and tyrosine metabolites are noted. Frequently, patients respond to supplemental ascorbic acid, a cofactor for pHPPD, and a lowered protein diet. Most affected infants are detected during newborn screening for PKU or tyrosinemia.
Alcaptonuria
Alcaptonuria, an autosomal recessive disorder, is associated with deficient activity of homogentisic acid oxidase and elevated levels of homogentisic acid. Homogentisic acid
polymerizes to form a pigment deposited in connective tissues throughout the body (ochronosis). Usually, pigment deposition is evident first in ear cartilage and sclera between ages 20 and 30 years. Later, deposition in the large joints and lumbosacral spine may lead to decreased range of motion and ankylosis. Acute inflammatory “ochronotic arthritis” may occur. Urine from affected patients may appear dark brown or may turn dark brown if left standing (i.e., autooxidize) or with alkalinization. If washed with alkaline solutions, cloth diapers will show dark-brown staining. A brownish discoloration may be noted in the axillary and inguinal areas of affected individuals. Perspiration will stain clothes. Elevated levels of homogentisic acid are noted in blood, urine, or tissue samples. Supplemental ascorbic acid may delay the onset of the disorder and reduce the severity of clinical symptoms.
polymerizes to form a pigment deposited in connective tissues throughout the body (ochronosis). Usually, pigment deposition is evident first in ear cartilage and sclera between ages 20 and 30 years. Later, deposition in the large joints and lumbosacral spine may lead to decreased range of motion and ankylosis. Acute inflammatory “ochronotic arthritis” may occur. Urine from affected patients may appear dark brown or may turn dark brown if left standing (i.e., autooxidize) or with alkalinization. If washed with alkaline solutions, cloth diapers will show dark-brown staining. A brownish discoloration may be noted in the axillary and inguinal areas of affected individuals. Perspiration will stain clothes. Elevated levels of homogentisic acid are noted in blood, urine, or tissue samples. Supplemental ascorbic acid may delay the onset of the disorder and reduce the severity of clinical symptoms.
DISORDERS OF HISTIDINE METABOLISM
Histidine, an essential amino acid in infants and children, is needed for nitrogen retention and growth. Histidine is metabolized through urocanic acid to formiminoglutamic acid and, ultimately, to glutamic acid.
Histidinemia
Histidinemia is associated with deficient activity of histidase (histidine ammonia lyase), which normally catalyzes the conversion of histidine to urocanic acid. This autosomal recessive condition occurs in approximately 1 in 12,000 births and results from mutations at the HAL gene locus at chromosome 12q22-q23. Affected patients have elevated plasma and urine levels of histidine. Increased urinary levels of histidine metabolites (i.e., imidazolepyruvic acid, imidazolelactic acid, and imidazoleacetic acid) produce a green color with the ferric chloride reagent, which can be confused with the blue-green color noted with phenylpyruvic acid in PKU. The majority of affected patients have no clinical effects from the elevated histidine levels. Approximately 1%, however, have psychomotor retardation and speech problems that suggest that the elevated histidine levels may play a role in the development of neurologic problems by enhancing the effects of other adverse clinical problems (i.e., perinatal hypoxia) at an early age. Special low-histidine diets will lower blood histidine levels and may be indicated in symptomatic patients. Deficient activity of histidase can be shown in liver or uncultured skin biopsies.
Urocanic Aciduria
Urocanic aciduria results from deficient activity of urocanase, which normally catalyzes the conversion of urocanic acid to imidazole propionic acid. Elevated levels of urocanic acid are found in the urine of affected patients. It remains unclear whether this finding has any clinical significance or is related to the psychomotor or growth retardation noted in some affected patients.
DISORDERS OF TRANSSULFURATION
The transsulfuration pathway is shown in Figure 385.3. Methionine, an essential amino acid, is converted to homocystine and subsequently is metabolized through cystathionine and cystine to inorganic sulfur, which is excreted from the body. Homocystine also may be remethylated to methionine by two pathways, which use either 5-methyltetrahydrofolate or betaine as methyl donors for the reaction. S-adenosylmethionine,
an intermediate in the conversion of methionine to homocystine, is an important donor for methyl groups needed for DNA and RNA modification and for reactions that involve neurotransmitter, myelin, and phospholipid metabolism. Because the demand for methyl groups often is greater than the amount that dietary methionine can supply, at least one-half of the homocystine produced from methionine is remethylated to methionine to meet this need.
an intermediate in the conversion of methionine to homocystine, is an important donor for methyl groups needed for DNA and RNA modification and for reactions that involve neurotransmitter, myelin, and phospholipid metabolism. Because the demand for methyl groups often is greater than the amount that dietary methionine can supply, at least one-half of the homocystine produced from methionine is remethylated to methionine to meet this need.
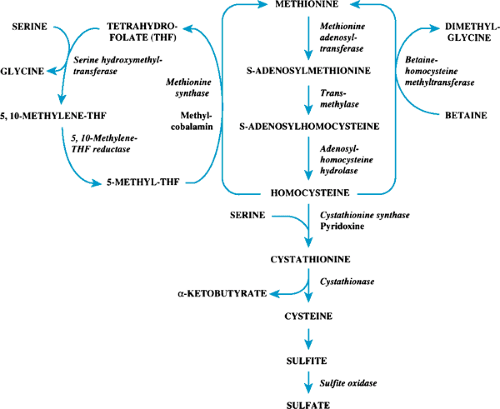 FIGURE 385.3. The transsulfuration pathway. |
Hypermethioninemia
Hypermethioninemia that occurs without elevation of other amino acids of the transsulfuration pathway results from deficient activity of methionine S-adenosyltransferase. No associated clinical manifestations are apparent. Most affected infants are detected by newborn screening programs that use measurement of blood methionine levels to detect homocystinuria. No specific treatment is needed. Elevated methionine levels may be seen also with severe liver dysfunction, hepatorenal tyrosinemia, and treatment with 6-azauridine or isoniazid.
Homocystinuria
Homocystinuria may occur as a result of cystathionine synthase deficiency. It may be the result also of defects in the remethylation pathway catalyzed by methionine synthase, which converts homocystine to methionine.
Cystathionine Synthase Deficiency
Classic homocystinuria is associated with deficient activity of cystathionine synthase. The disorder is inherited as an autosomal recessive trait and has an incidence of approximately 1 in 344,000 births. Elevated levels of methionine and homocystine are found in the body fluids of affected patients. Cystine levels are low. Affected patients appear normal at birth, then slowly develop clinical features of the disease during early childhood. As in hypermethioninemia, the elevated methionine levels appear to be benign. The major clinical manifestations are related to the elevated homocystine levels. Interference in the cross-linkage of collagen leads to dislocated lens, osteoporosis, scoliosis, and a marfanoid body habitus. Most patients are tall, with long, thin extremities. In contrast to Marfan syndrome, often they have decreased range of motion at the elbows and knees and develop pes cavus foot deformities (Fig. 385.4). Elevated homocystine levels also disrupt the vascular endothelium, and thrombus formation can occur in any vessel at any age. Hypertension, pulmonary emboli, myocardial infarctions, and cerebrovascular accidents occur. Approximately 25% of patients have a major thromboembolic event by age 20 years; 50% have a major event by age 28. Psychomotor retardation develops; the average intelligence quotient is 64. Unusual personalities, behavioral problems, and other neuropsychiatric problems are common.
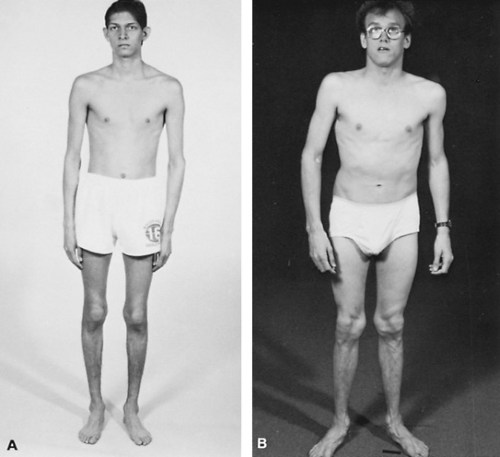 FIGURE 385.4. Homocystinuria. A: Teenage boy who presented with a pulmonary embolus. B: Young adult. |
Plasma and urine quantitative amino acid determinations will show elevated levels of methionine and homocystine and lowered levels of cystine. Other sulfur-containing amino acids may be noted (i.e., the mixed disulfide of cysteine and homocysteine, homolanthionine, and S-adenosyl homocysteine). Protein-bound homocystine levels will be elevated. Deficient activity of cystathionine synthase may be shown in phytohemagglutinin-stimulated lymphocytes or in cultured skin fibroblasts.
Treatment is aimed at reducing homocystine levels to as nearly undetectable as possible and at normalizing cystine levels. Limited amounts of natural foods and special methionine-free medical foods with added cystine are given. Betaine, which serves as a methyl donor in the remethylation of homocystine to methionine, is used in addition to dietary therapy. Supplemental vitamins may be needed to ensure adequate intake of
pyridoxine, vitamin B12, and folate. Although treatment is indicated for life, achieving compliance with dietary therapy in adolescents and adults may be difficult.
pyridoxine, vitamin B12, and folate. Although treatment is indicated for life, achieving compliance with dietary therapy in adolescents and adults may be difficult.
Approximately 50% of patients with homocystinuria have some residual cystathionine synthase activity and are responsive to high-dose pyridoxine therapy. Usually, these patients have less severe clinical manifestations of the disorder. Many have normal intelligence. Doses of 300 to 900 mg/day of pyridoxine may be given in adults. Doses in children should be proportionately lower and not exceed 900 mg/1.73 m2/day. Special medical foods and betaine therapy may be needed in some patients for control of the disorder. Others require only a relatively lowered natural protein diet.
The gene that encodes for cystathionine synthase (CBS) is located at chromosome 21q22.3. At least 92 mutations in the CBS gene have been reported in affected patients, some of which are associated with pyridoxine responsiveness. Carrier detection may be performed if the mutation or polymorphisms at the CBS locus are known. Prenatal diagnosis is available.
Newborn screening for homocystinuria occurs in some states and in Europe by testing blood methionine levels in dried blood filter-paper samples. In many affected infants the disorder has not been detected, however, mainly owing to the slow rise in blood methionine levels during the first few days of life when screening samples are customarily obtained in the United States. In those infants with homocystinuria who were detected by newborn screening and who were started on dietary therapy in early infancy, the natural course of the disease has been modified. Psychomotor handicaps and dislocated lenses have been prevented or delayed in onset. With the use of more sensitive newborn screening methods such as tandem mass spectrometry, which many states are now adopting, it is anticipated that improved early detection of homocystinuria patients will occur.
Homocystinuria Due to Remethylation Defects
Homocystinuria may result from defects in the remethylation pathway. Affected patients have deficient activity of methionine synthase or inherited disorders in folate or cobalamin metabolism that affect methionine synthase activity. Homocystine levels are elevated but to a degree less than that seen with cystathionine synthase deficiency. Methionine levels are low or normal. Cystathionine levels may be elevated. Usually, progressive central nervous system involvement occurs in addition to features of classic homocystinuria. All the disorders are inherited as autosomal recessive traits. Prenatal diagnosis is available. Carrier detection may be performed if the mutation or polymorphisms at the genetic loci involved are known. Nutritional deficiencies of folate or cobalamin may result in similar biochemical and clinical abnormalities.
Deficient Activity of Methionine Synthase
Methionine synthase (5-methyltetrahydrofolate homocysteine S-methyltransferase) catalyzes the remethylation of homocystine to methionine. Patients with the disorder present most often as infants with poor feeding, lethargy, hypotonia, delayed development, and megaloblastic anemias. One adult patient presented with neurologic problems and macrocytic anemia. Serum vitamin B12 and folate levels are normal. Some patients respond to treatment with intramuscular hydroxocobalamin, betaine, and folic acid. The disorder results from mutations in the gene MTF, located at chromosome 1q43, which encodes for methionine synthase. Previously, patients with methionine synthase deficiency were designated as having type cblG (cobalamin G) defects (named after the complementation groups noted in studies with cell lines of patients with various defects of cobalamin metabolism).
Defects in Tetrahydrofolate Metabolism
Deficient activity of 5,10-methylenetetrahydrofolate reductase (MTHFR) results in lowered levels of 5-methyltetrahydrofolate, the major transport form of folate, and the methyl donor for the methionine synthase reaction. The disorder may present at any age; clinical severity is related to the amount of residual enzyme activity present. The patients most severely affected are neonates who present with apnea, seizures, and a progressive demyelinating encephalopathy. Most do not survive beyond age 1 year. Other patients less severely affected have developmental delay, incoordination, gait abnormalities, paresthesias, seizures, recurrent strokes, or psychiatric problems. Some have a marfanoid body habitus. Megaloblastic anemias do not occur. Serum folate levels are normal or low; vitamin B12 levels are normal. Perivascular changes, demyelination, macrophage infiltration, and astrogliosis of the central nervous system occur in addition to thrombus formation as seen with cystathionine synthase deficiency. Patients with milder forms may respond to treatment with betaine, folic acid, pyridoxine, and hydroxocobalamin. Methionine supplementation also may be needed. Residual neurologic impairments may improve slowly with treatment and rehabilitation.
Deficient activity of MTHFR may be shown in peripheral blood leukocytes or cultured skin fibroblasts. A number of mutations have been reported in the gene MTHFR located at chromosome 1p36.3, which encodes for the enzyme. The disorder is inherited as an autosomal recessive trait. Carrier detection is available for families in whom the mutation or polymorphisms are known. Prenatal diagnosis is available.
A specific allele at the MTHFR locus (C677T) encodes for a thermolabile enzyme. Homozygosity for this allele, if combined with low tissue folate levels, may result in elevated plasma homocystine (hyperhomocystinemia) and an increased risk for atherosclerotic vascular disease. The allele also may play a role in the prevention of neural tube birth defects by folic acid.
Defects in Cobalamin Metabolism
Dietary cobalamin (vitamin B12) is taken up into the cells as hydroxocobalamin, is processed in lysosomes, and then is further metabolized to either methylcobalamin, a cofactor for methionine synthase, or adenosylcobalamin, a cofactor for methylmalonyl-CoA mutase. Depending on the site of the metabolic block, patients with defects in cobalamin metabolism may have only homocystinuria, only methylmalonic acidemia, or combined homocystinuria and methylmalonic acidemia. Table 385.1 lists the biochemical genetic features of the disorders of cobalamin metabolism along with classic homocystinuria and methylmalonic acidemia. The disorders have been classified into seven complementation groups, cblA to cblG, according to studies in cell lines from affected patients.
Deficient activity of methionine synthase reductase
Functional deficiency of methionine synthase results from defects in the formation of its cofactor methylcobalamin. Patients with type cblE complementation group have methionine synthase reductase deficiency as a result of mutations in the MTRR gene located at chromosome 5p15.3-p15.2. This defect in the formation of methylcobalamin occurs intracellularly after lysosomal processing of hydroxocobalamin. Affected patients present in infancy with clinical findings similar to those seen in methionine synthetase deficiency, including poor feeding, lethargy, hypotonia, and delayed development. They have megaloblastic anemias but do not have methylmalonic acidemia. Serum folate and vitamin B12 levels are normal. Most respond to treatment with large doses of intramuscular hydroxocobalamin. Betaine and folic acid also may be needed. One patient with cblE disease was detected and treated prenatally by maternal intramuscular hydroxocobalamin injections. This patient continues to do well on hydroxocobalamin therapy.
Abnormalities in cobalamin metabolism and activation that affect the formation of both methylcobalamin and adenosylcobalamin result in combined homocystinuria and methylmalonic acidemia.
Patients have reduced activities of methionine synthase and methylmalonyl-CoA mutase and clinical features of both associated disorders. Three complementation groups—cblC, cblD, and cblF—are associated with these abnormalities. Type cblF results from a defect in the transfer of hydroxocobalamin from lysosomes to the cytosol. The exact intracellular metabolic defects in types cblC and cblD are unknown. Most affected patients present in infancy with failure to thrive, poor feeding, lethargy, hypertonicity, and progressive neurologic dysfunction. Cortical atrophy and myelopathies may be seen, especially in those patients with later onset of their disease. Pigmentary retinopathies and abnormal maculae have been reported with types cblC and cblD. Type cblC may present with hemolytic-uremic syndrome. One case of sudden death was reported with type cblF. Some older patients have a marfanoid habitus. Megaloblastic or megalocytic anemias and, occasionally, pancytopenia occur. Serum cobalamin and folate levels are normal. Moderately elevated homocystine and normal or lowered methionine levels are noted on quantitative amino acid determinations. Urine organic acid analysis will show moderate to marked elevation of methylmalonate but less than that seen in classic methylmalonic acidemia. Usually, severe metabolic acidosis does not occur. Some patients respond to treatment with large doses of intramuscular hydroxocobalamin, betaine, and diets lower in natural protein. Psychomotor handicaps, however, may not be reversible. Others do not respond to therapy and die in infancy. Prenatal diagnosis is available.
Patients have reduced activities of methionine synthase and methylmalonyl-CoA mutase and clinical features of both associated disorders. Three complementation groups—cblC, cblD, and cblF—are associated with these abnormalities. Type cblF results from a defect in the transfer of hydroxocobalamin from lysosomes to the cytosol. The exact intracellular metabolic defects in types cblC and cblD are unknown. Most affected patients present in infancy with failure to thrive, poor feeding, lethargy, hypertonicity, and progressive neurologic dysfunction. Cortical atrophy and myelopathies may be seen, especially in those patients with later onset of their disease. Pigmentary retinopathies and abnormal maculae have been reported with types cblC and cblD. Type cblC may present with hemolytic-uremic syndrome. One case of sudden death was reported with type cblF. Some older patients have a marfanoid habitus. Megaloblastic or megalocytic anemias and, occasionally, pancytopenia occur. Serum cobalamin and folate levels are normal. Moderately elevated homocystine and normal or lowered methionine levels are noted on quantitative amino acid determinations. Urine organic acid analysis will show moderate to marked elevation of methylmalonate but less than that seen in classic methylmalonic acidemia. Usually, severe metabolic acidosis does not occur. Some patients respond to treatment with large doses of intramuscular hydroxocobalamin, betaine, and diets lower in natural protein. Psychomotor handicaps, however, may not be reversible. Others do not respond to therapy and die in infancy. Prenatal diagnosis is available.
TABLE 385.1. BIOCHEMICAL GENETIC FEATURES OF METHYLMALONIC ACIDEMIA AND HOMOCYSTINURIA | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
Cobalamin metabolism abnormalities that affect only the formation of adenosylcobalamin, complementation groups cblA and cblB, result in methylmalonic acidemia. They are discussed with that disorder.
Cystathioninuria
Elevated levels of cystathionine and related sulfur-containing compounds, from cystathioninase (cystathionase) deficiency, are benign and not associated with any significant clinical manifestations. The elevated levels of cystathionine may normalize with pyridoxine treatment.
Increased levels of cystathionine may be seen also transiently in normal neonates. They also may occur with nutritional pyridoxine deficiency, thyrotoxicosis, hepatic dysfunction, and certain tumors (i.e., neuroblastomas, ganglioblastomas, and hepatomas).
Sulfite Oxidase Deficiency
Sulfite oxidase deficiency is a rare autosomal recessive disorder that results in the accumulation and increased urinary excretion of sulfite, thiosulfate, and S-sulfocysteine. Affected patients present as neonates with severe, generalized, intractable seizures. Dislocated lens and a demyelinating central nervous system deterioration occur in those who survive the neonatal period.
Deficiency of the molybdenum cofactor for sulfite oxidase deficiency is more common than is isolated sulfite oxidase deficiency and presents with similar clinical findings. Molybdenum is a cofactor also for xanthine dehydrogenase and aldehyde oxidase. Affected patients have low levels of uric acid; elevated levels of urinary xanthine, hypoxanthine, and taurine; and accumulation of sulfite, thiosulfate, and S-sulfocysteine. Milder variants have been reported. Deficient activity of sulfite oxidase
or xanthine dehydrogenase may be demonstrated in cultured skin fibroblasts or liver biopsy samples. No specific treatment is available. Prenatal diagnosis is available.
or xanthine dehydrogenase may be demonstrated in cultured skin fibroblasts or liver biopsy samples. No specific treatment is available. Prenatal diagnosis is available.
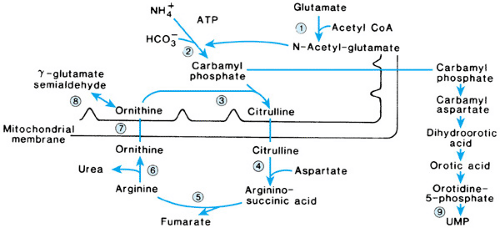 FIGURE 385.5. Urea cycle. 1, N-acetylglutamate synthetase; 2, carbamyl phosphate synthetase I; 3, ornithine transcarbamylase; 4, argininosuccinate synthetase; 5, argininosuccinate lyase; 6, arginase; 7, mitochondrial ornithine transport defect (hyperammonemia–hyperornithinemia–homocitrullinuria); 8, ornithine aminotransferase; 9, decarboxylase, site of allopurinol block in pyrimidine pathway. ATP, adenosine triphosphate; CoA, coenzyme A; UMP, uridine monophosphate. |
DISORDERS OF AMMONIA METABOLISM
Excess dietary or waste nitrogen (remaining after what is needed for protein synthesis and tissue maintenance is used) normally is not stored in the body but is converted into urea by a series of reactions known as the urea cycle (Fig. 385.5). Disorders of the urea cycle are associated with the accumulation of ammonia and its precursors (i.e., glutamine, glutamic acid, aspartic acid, and glycine). Elevated plasma ammonia levels that exceed three times the upper limits of normal are toxic and associated with cytotoxic changes in the brain and liver. With rising ammonia levels, affected patients develop poor feeding, anorexia, behavioral changes, irritability, vomiting, lethargy, ataxia, and seizures. As the hyperammonemia progresses, affected patients become comatose, and ventilatory support may be needed. Circulatory collapse and cerebral edema may occur. The classic cases are neonates who appear asymptomatic for 24 to 48 hours and then develop progressive neurologic deterioration. Milder forms of the disorders may present later in infancy, may produce intermittent symptoms over a period of years, or may be detected in older children or adults with neurologic problems or psychomotor retardation. As a group, the disorders are estimated to occur in approximately 1 in 30,000 live births. All the disorders of ammonia metabolism are inherited as autosomal recessive traits, except for ornithine transcarbamylase (OTC) deficiency, which is inherited as an X-linked trait, and transient hyperammonemia of the newborn, which is not genetic in origin.
The diagnosis of a specific urea cycle disorder is made on the basis of the pattern of plasma and urine amino acid abnormalities and the presence or absence of orotic aciduria. Confirmation of specific enzymatic deficiencies requires only erythrocytes or cultured skin fibroblasts for some of the disorders but necessitates liver biopsy for others. Molecular genetic studies may be used also with many of the disorders to confirm the diagnosis and for carrier and prenatal testing.
Secondary hyperammonemia, associated with other amino acidemias or organic acidurias, usually can be excluded by the absence of acidosis. Urinary organic acid determination, however, should be obtained to rule out the rare case that might present initially with hyperammonemia (i.e., with maple syrup urine disease [MSUD], methylmalonic acidemia, or propionic acidemia). Secondary hyperammonemia may occur also with disorders of mitochondrial fatty acid oxidation or oxidative phosphorylation and with severe liver dysfunction or liver failure.
Treatment of acute hyperammonemia, either at the initial presenting episode or at a subsequent intercurrent episode, is a medical emergency. With the initial episode, blood and urine samples should be collected for diagnostic testing, but treatment should be started immediately, before a specific diagnosis is established. Therapy should include the removal of all exogenous protein sources and the administration of intravenous glucose to prevent protein catabolism. Reduction of markedly elevated plasma ammonia and ammonia precursors is carried out most effectively by hemodialysis. Alternatively, hemofiltration may be used. For a temporary delay in hemodialysis or hemofiltration, peritoneal dialysis may be performed but is less effective. Because the duration of time in coma is inversely related to outcome, prompt referral to a tertiary medical center is indicated if hemodialysis or hemofiltration are not readily available.
Drugs that use alternate pathways for waste nitrogen excretion, such as sodium benzoate and sodium phenylacetate, may be used intravenously to control acute mild to moderate hyperammonemia (less than 350 μmol). One must be prepared to start hemodialysis or hemofiltration immediately in the absence of significant improvement in ammonia levels within a short period after this therapy is instituted, however. These drugs are used enterally also to maintain plasma ammonia levels within the normal range between intercurrent episodes in patients with the more severe forms of the disorders. Sodium benzoate combines with glycine to form hippurate. Sodium phenylacetate combines with glutamine to form phenylacetylglutamine. Both hippurate and phenylacetylglutamine are excreted rapidly by the kidneys and remove a significant amount of nitrogen, the ammonia precursor. A combination preparation of sodium benzoate and sodium phenylacetate is commercially available (previously investigational new drug) for intravenous use. Sodium phenylbutyrate, which is converted into sodium phenylacetate in the body and is less odorous than phenylacetate, also is commercially available in tablet or powder form for enteral use. Before these drugs were developed, many patients with severe forms of the disorders did not survive past age 1 year.
Once plasma ammonia levels fall to less than 100 μmol, the patient may be started on enteral therapy. Special medical foods, free of protein or with protein content composed of only essential amino acids, may be necessary to meet basic caloric and other nutrient needs. Depending on the type of disorder and its severity, a fairly limited protein intake with or without essential amino acid supplementation will be needed. Specific amino acid supplementation with L-citrulline or L-arginine also is given for relative insufficiencies of these amino acids and to “prime” the urea cycle. Intravenous L-arginine hydrochloride
is used for the same purpose during acute hyperammonemic episodes.
is used for the same purpose during acute hyperammonemic episodes.
Even with prompt and aggressive medical therapy, only 30% to 50% of neonates who develop hyperammonemic coma survive the neonatal period. Most of those who do survive have significant neurologic deficits and psychomotor retardation. Seizure disorders, cortical atrophy, and spastic quadriparesis are common. Later acute episodes, usually precipitated by intercurrent infections or excessive protein intake, also may lead to further neurologic sequelae or death. Liver transplantation, done before significant neurologic impairment has occurred, has been shown to correct the biochemical abnormalities in children affected with severe forms of the disorders.
Carbamyl Phosphate Synthetase I Deficiency
Carbamyl phosphate synthetase I (CPS I) is a mitochondrial enzyme that catalyzes the formation of carbamyl phosphate from ammonia, adenosine triphosphate, and bicarbonate in the presence of N-acetylglutamate. N-Acetylglutamate, a critical cofactor for this reaction, is formed from acetyl CoA and glutamate in the presence of N-acetylglutamate synthetase. Inhibition of N-acetylglutamate synthetase by organic acids, especially propionic acid, is thought to be the reason for the secondary hyperammonemia seen with organic acidurias.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


