Diseases of the Neuromuscular Junction
Julie Thorne Parke
Numerous conditions may interfere with the transmission of the electrical impulse across the neuromuscular junctions, which consist of the terminal portion of a motor nerve, the synaptic cleft, and the end-plate region of a muscle. The nerve impulse originates in the anterior horn cell and is propagated down the axon of the motor nerve into the motor nerve terminals. Depolarization of the nerve terminals opens calcium channels, causing the release of acetylcholine into the synaptic cleft. Acetylcholine binds to receptors on the muscle end plate, altering its permeability to ions and causing localized depolarization of the end plate (the end-plate potential). If the amplitude of the end-plate potential reaches threshold, a muscle fiber action potential is generated. The muscle action potential is propagated along the muscle fiber and into the interior of the muscle fiber by the T tubules, thus initiating muscle fiber contraction. Acetylcholine acts at the postsynaptic membrane for only a brief period before it is broken down by an enzyme, cholinesterase, into two inactive components, choline and acetic acid. The choline is taken up by the presynaptic nerve terminal, where choline acetyltransferase catalyzes the resynthesis of acetylcholine.
Neuromuscular transmission can fail if insufficient acetylcholine is released (presynaptic process) or if the number of acetylcholine receptors is insufficient to interact with the acetylcholine (postsynaptic disorder). Conditions interfering with the presynaptic events include some forms of congenital myasthenia gravis, botulism, hypocalcemia, hypermagnesemia, and neuromuscular blockade from antibiotics. Disorders affecting the postsynaptic events include autoimmune myasthenia gravis, some types of congenital myasthenia gravis, organophosphate poisoning, and iatrogenic neuromuscular blockade with curare (Table 408.1). Neuromuscular transmission failure also may occur when inhibition of or a deficiency in acetylcholinesterase occurs, causing a depolarization block.
Disorders of neuromuscular transmission are manifested clinically by muscle weakness, which is exacerbated by exercise and improved by rest. Defects in neuromuscular transmission can be documented by pharmacologic tests and by electrophysiologic studies, including repetitive nerve stimulation and single-fiber electromyography.
JUVENILE MYASTHENIA GRAVIS
Pathophysiology
The juvenile and adult forms of myasthenia gravis are autoimmune disorders characterized by an autoimmune attack on the acetylcholine receptor. Circulating antibodies to the acetylcholine receptor bind to the receptor on the muscle end plate, blocking its function. Morphologic studies show a simplified postsynaptic membrane with poorly developed folds and clefts and a loss of functional acetylcholine receptor sites. Antibody can be demonstrated on the postsynaptic membrane, further implicating an immunologic process in its destruction. Circulating acetylcholine receptor antibodies can be measured, but the titer does not correlate well with the clinical condition of affected patients. A lymphocyte-mediated immune response to acetylcholine receptors also has been identified. The thymus plays a role in the disease, possibly by sensitizing specific lymphocytes to produce acetylcholine receptor antibodies.
Clinical Manifestations and Complications
Usually, the onset of juvenile myasthenia gravis occurs after age 10, although it can appear much earlier. Girls are affected more commonly than are boys. The cardinal feature of the disease is easy fatigability. Usually, the onset is gradual, with symptoms most apparent in the afternoon or evening, when the patient is tired. Occasionally, the onset is fairly sudden and may appear to have been precipitated by an infectious illness. Characteristically, the weakness abates with rest and worsens with sustained effort. In approximately one-half of patients, weakness first appears in the ocular muscles, causing ptosis or diplopia (Fig. 408.1). Frequently, ptosis is asymmetric and may be unilateral. It tends to fluctuate during the day and to vary from day to day. Involvement of the ocular muscles is variable, but it may be severe, causing a total ophthalmoplegia. Approximately one-fourth of affected patients have weakness of the bulbar musculature, resulting in difficulties in speaking, swallowing, or chewing. The facial muscles are involved in most affected patients. Weakness of the palate and
tongue may render speech unintelligible. Affected children’s voices may be strong initially, becoming softer and less distinct during continued conversation. Difficulty chewing food is a common problem, and many patients support their jaw in one hand to assist with chewing. Swallowing difficulties and choking spells may occur. Weakness of the muscles of the neck, particularly the neck extensors, causes the head to fall forward. Patients with predominantly bulbar symptoms are at risk of developing respiratory failure, particularly during an intercurrent infection.
tongue may render speech unintelligible. Affected children’s voices may be strong initially, becoming softer and less distinct during continued conversation. Difficulty chewing food is a common problem, and many patients support their jaw in one hand to assist with chewing. Swallowing difficulties and choking spells may occur. Weakness of the muscles of the neck, particularly the neck extensors, causes the head to fall forward. Patients with predominantly bulbar symptoms are at risk of developing respiratory failure, particularly during an intercurrent infection.
TABLE 408.1. DISORDERS OF NEUROMUSCULAR TRANSMISSION | |
|---|---|
|
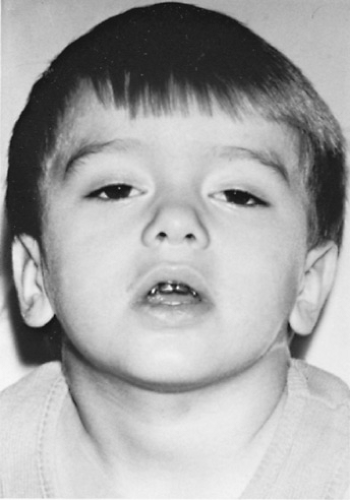 FIGURE 408.1. Four-year-old child with juvenile myasthenia gravis, exhibiting fluctuating ptosis and bilateral facial weakness. |
A smaller number of children (approximately 20%) have generalized weakness of the extremities. Fatigability may be demonstrated in younger children by having them climb stairs or hold their arms outstretched for an interval. In older children, repetitive testing of deltoid strength or performance of multiple deep knee bends may help to disclose the weakness. Regardless of the distribution of weakness, the principal features are a fluctuating quality in the weakness and a susceptibility to fatigue. These features differ from those of other neuromuscular disorders, which produce relatively constant symptoms.
Diagnosis
Usually, the diagnosis of myasthenia gravis can be made on the basis of the history and physical examination, and it may be confirmed by pharmacologic tests. A small dose of an anticholinesterase drug produces a dramatic improvement in strength. Edrophonium chloride (Tensilon) is preferred because of its rapid onset and short duration of action. The availability of acetylcholine is increased by inhibiting the enzyme cholinesterase, thereby improving neuromuscular transmission. A placebo injection of normal saline should be given before the edrophonium. A test dose of one-tenth of the total dose is given initially. If no complications occur with the test dose, the remainder of the full dosage of 0.1 mg/kg (maximum dosage, 10 mg) is given intravenously. The affected patient’s heart rate and blood pressure must be monitored throughout the test, and atropine sulfate should be immediately available because a cholinergic crisis occasionally occurs, manifest by extreme bradycardia or transient respiratory weakness requiring ventilatory support. Usually, a marked but short-lived improvement in weakness is seen in patients with myasthenia. A more prolonged effect can be achieved with intramuscular or subcutaneous Neostigmine if a longer observation period is necessary to evaluate limb strength. Neostigmine is especially useful in infants and younger children.
Electrophysiologic studies are helpful in documenting transmission failure at the neuromuscular junctions. Repetitive nerve stimulation produces a characteristic fall in amplitude between the first and the fourth or fifth responses (decremental response). Testing several muscles may be necessary because the abnormality may not be present in all muscles. Selective single-fiber electromyography is possible in some older patients and may confirm the variability in synaptic transmission time in patients with rather mild disease. Antibodies to the human muscle acetylcholine receptor are found in the serum of as many as 90% of patients. However, patients with negative antibody test results are those who typically present with a purely ocular weakness or mild generalized weakness and in whom the diagnosis is uncertain. A negative test result does not exclude the diagnosis.
Therapy and Prognosis
Numerous different therapeutic modalities are available for treating myasthenia gravis. The selected approach should consider the age of the affected patient, the severity of the disease, and the potential benefits and risks of each form of therapy. Cholinesterase inhibitors improve neuromuscular transmission by inhibiting the enzymatic degradation of acetylcholine and thereby prolonging its effect on muscle end plates. These agents result in symptomatic improvement in strength in most patients with myasthenia gravis and may be sufficient to produce normal or near-normal strength in some. Pyridostigmine bromide (Mestinon) and neostigmine bromide (Prostigmin) are the agents used most commonly. Pyridostigmine is preferred because of its longer half-life and more favorable side-effect profile. An initial dosage of 1.0 mg/kg/day in divided dosages is typical in pediatric patients. Adolescents and adults typically are started at dosages of 30 to 60 mg every 4 to 6 hours. The dosage and the dosing interval must be adjusted carefully on the basis of close clinical observation. The dosage required by given individuals may vary during the day and from one day to the next. A cholinergic crisis may result from excessive anticholinesterase dosing, as a result of the accumulation of acetylcholine at neuromuscular junctions. Both nicotinic symptoms, such as increased muscle weakness and fasciculations, and muscarinic symptoms, such as diarrhea, pallor, sweating, increased salivation, cardiovascular disturbances, and visual blurring, may occur (Table 408.2). Clinicians may have difficulty in distinguishing between an overdose of anticholinesterase medications (producing weakness in respiratory muscles) and respiratory distress from myasthenic crisis (causing respiratory insufficiency). Close monitoring of the affected patient’s muscle strength, pulmonary function, and ability to cough adequately is critical during these periods. Elective intubation and ventilatory support should be instituted before respiratory insufficiency occurs.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


