Essentials of Diagnosis
- Symmetric proximal muscle weakness progressing over weeks to months.
- Elevated muscle enzymes, including creatine kinase (CK), aldolase, aspartate aminotransferase (AST), and alanine aminotransferase (ALT).
- An “irritable myopathy” shown by electromyography (EMG).
- MRI of affected muscles reveals evidence of edema or fasciitis or both.
- A heliotrope rash, Gottron sign/papules are pathognomonic for dermatomyositis.
- Muscle biopsy findings frequently reveal endomysial, perimysial, and perivascular lymphocytic infiltrates. Except for perifascicular atrophy, which is pathognomonic for dermatomyositis, muscle biopsy findings are variable and nonspecific.
- A careful family history, medication list review, physical examination, laboratory evaluation, and muscle biopsy are critical and help exclude an alternative diagnosis, such as an inherited muscle disease or toxic myopathy.
General Considerations
Polymyositis and dermatomyositis are a rare, heterogeneous group of autoimmune myopathies with an approximate incidence of 1 case per 100,000 per year. Although polymyositis is virtually unheard of in children, juvenile dermatomyositis is well-described and occurs most frequently between the ages of 10 and 15 years. In adults, the autoimmune myopathies can occur at any age but seem to peak between the ages of 45 and 60 years.
In polymyositis and dermatomyositis, muscle biopsies are characterized by lymphocytic infiltrates. Recently, however, a form of autoimmune muscle disease with prominent myofiber degeneration and necrosis with minimal, if any, inflammatory cells has also been recognized. This form of myositis, termed “immune-mediated necrotizing myopathy,” has been included in the most contemporary diagnostic classification schemes of the autoimmune myopathies.
Although most patients with dermatomyositis have both skin and muscle involvement, patients occasionally have only the skin manifestations and are classified as having “dermatomyositis sine myositis” or amyopathic dermatomyositis. Conversely, in rare instances, the classic muscle biopsy features of dermatomyositis are observed in the absence of rash. These patients are said to have “dermatomyositis sine dermatitis” (Table 27–1).
|
Clinical Findings
Symmetric proximal muscle weakness evolving over weeks to months is the presenting symptom in most patients. Typical complaints include difficulty rising from a low chair, walking up steps, and washing one’s hair. In more severe cases, weakness of the neck flexors, pharyngeal weakness, and diaphragmatic weakness can cause head drop, dysphagia, and respiratory compromise, respectively. On physical examination, weakness of the proximal arm muscles, especially the deltoids, but often including the biceps and triceps, is expected. Hip flexors are the most commonly affected leg muscles but the hamstrings and quadriceps are also frequently weak. Subtle leg weakness can be detected by having the patient attempt to rise from a 6-inch high stool without using the arms.
As a general rule in the autoimmune myopathies, distal weakness should only occur in the presence of severe proximal muscle weakness. Isolated or even mild distal weakness should always raise doubts about the diagnosis of dermatomyositis, polymyositis, or immune-mediated necrotizing myopathy. The examiner can test whether distal weakness is present by evaluating the strength of wrist flexors, wrist extensors, distal finger flexors, and finger extensors. In the lower extremities, ankle dorsiflexion and ankle plantarflexion strength should be assessed manually and by having the patient attempt to walk on their heels and toes. The presence of facial weakness or scapular winging (Plates 43 and 44) should be assessed, since these are extremely atypical in autoimmune myopathy and suggest the probability of an alternative diagnosis (see Differential Diagnosis section).
In addition to muscle weakness, arthralgias or frank arthritis, myalgias, severe fatigue, Raynaud phenomenon, or symptoms of another overlapping rheumatologic condition (such as lupus or scleroderma) may be present. Dyspnea may reflect diaphragmatic weakness or, especially in patients with the antisynthetase syndrome (see below), interstitial lung disease. The latter is often associated with a persistent dry cough, crackles on chest auscultation, and decreased oxygen saturation with exercise. Serious clinically significant cardiac manifestations occur, albeit rarely, in the autoimmune myopathies.
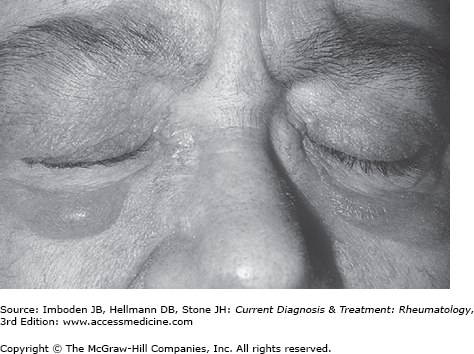
Patients with dermatomyositis may present with cutaneous manifestations either before or after the development of muscle symptoms. Gottron papules are raised violaceous lesions at the extensor surfaces of the metacarpophalangeal, proximal interphalangeal, and the distal interphalangeal joints (Figure 27–1). The Gottron sign is an erythematous rash involving these sites that can also be found at the extensor surfaces of the elbows and knees. The heliotrope rash is an often red or purplish discoloration of the eyelids (Figure 27–2); in blacks, this may appear hyperpigmented. Although both the heliotrope and Gottron rashes are pathognomonic for dermatomyositis, other less specific rashes may occur. These include an erythematous or poikilodermatous rash across the posterior neck and shoulders (the shawl sign) and a similar rash on the anterior neck and chest (the V-sign). In some patients, dermatomyositis-associated rashes are sun-sensitive. In some patients, particularly those with the antisynthetase syndrome (see below), hyperkeratotic skin thickening, often with painful cracking, on the radial surfaces of the fingers (mechanic’s hands) or toes (mechanic’s feet) may develop. In addition, periungual telangiectasias and nailfold capillary changes identical to those found in scleroderma may develop.
Figure 27–1.
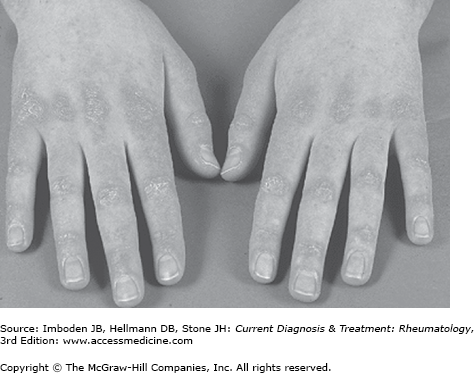
Dermatomyositis. Gottron papules on the dorsa of the hands and fingers, especially over the metacarpophalangeal and interphalangeal joints. (Reproduced, with permission, from Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. McGraw-Hill, 2009.)
In general, no definite environmental exposures have been associated with the development of polymyositis. While increased UV light exposure may predispose patients to dermatomyositis, a history of intense sun exposure is only occasionally elicited from patients with this condition. In contrast, there is emerging evidence that a distinct form of immune-mediated necrotizing myopathy may be triggered by statin use and is associated with autoantibodies recognizing HMG-CoA reductase, the pharmacologic target of statins (see below).
Creatine kinase (CK), aldolase, AST, ALT, and lactate dehydrogenase, are released from damaged muscle and elevated levels are often, but not always, found in patients with autoimmune myopathy. In some instances of dermatomyositis and polymyositis, the aldolase may be elevated in the presence of a normal CK level. Not infrequently, elevated AST and ALT levels are misinterpreted as evidence of liver disease in patients with myopathy. To rule out liver disease, a serum gamma glutamyl transferase (GGT) level can be measured; GGT is usually released along with AST and ALT in liver disease but not from damaged muscle.
As in other systemic autoimmune diseases, there is a strong association of autoantibodies against specific autoantigens with distinct clinical phenotypes. The term “myositis-specific autoantibody” has been used to describe those antibodies that are found in ˜60–80% of patients with polymyositis, dermatomyositis, and immune-mediated necrotizing myopathy but not other rheumatic diseases or neuromuscular disorders (Table 27–2).
| Name | Antigen | Clinical Manifestation |
|---|---|---|
| Antisynthetase autoantibodies | ||
| Anti-Jo-1 | Histidyl t-RNA synthetase | PM or DM with ILD |
| Anti-PL-7 | Threonyl t-RNA synthetase | PM or DM with ILD |
| Anti-PL-12 | Alanyl t-RNA synthetase | ILD more often than Myo |
| Anti-EJ | Glycyl t-RNA synthetase | PM more often than DM with ILD |
| Anti-OJ | Isoleucyl t-RNA synthetase | ILD with PM/DM |
| Anti-KS | Asparaginyl t-RNA synthetase | ILD more often than Myo |
| Anti-Zo | Phenylalanyl t-RNA synthetase | ILD with Myo |
| Anti-Ha | Tyrosyl t-RNA synthetase | ILD with Myo |
| Nonsynthetase autoantibodies | ||
| Dermatomyositis-specific autoantibodies | ||
| Anti-Mi-2 | DNA helicase | Dermatomyositis with rash > muscle symptoms, treatment responsive |
| Anti-MDA5 (anti CADM 140) | Melanoma differentiation-associated gene 5 | DM: CAM, DM with rapidly progressive lung disease, pneumomediastinum |
| Anti-155/140 | Transcriptional intermediary factor 1-gamma | CAM |
| Anti-140 | Nuclear matrix protein (NXP-2) | Juvenile DM |
| Anti-SAE | Small ubiquitin-like modifier-activating enzyme | DM: CAM, DM with rapidly progressive lung disease, pneumomediastinum |
| Immune-mediated necrotizing myopathy–specific autoantibodies | ||
| Anti-SRP | Signal recognition particle | Severe, acute, resistant necrotizing myopathy |
| Anti-HMGCR (anti 200/100) | HMG CoA Reductase | Necrotizing myopathy related to statin use in majority. Majority of patients received statin therapy but also reported in a minority of patients who did not receive statin therapy |
Autoantibodies recognizing one of the aminoacyl tRNA synthetases (eg, anti-Jo-1) are the most common of the myositis-specific autoantibodies and occur in ˜30% of patients with polymyositis or dermatomyositis. In addition to an autoimmune myopathy, patients with antisynthetase autoantibodies may have one or more of the following features: interstitial lung disease, nonerosive arthritis, fevers, Raynaud phenomenon, and mechanic’s hands. Patients with 1 of the antisynthetase autoantibodies and 2 or more of these features are said to have the antisynthetase syndrome. Interestingly, different antisynthetase antibodies appear to be associated with these features at different frequencies. For example, in patients with anti-Jo-1, 90% have muscle involvement and 50–75% have interstitial lung disease. In contrast, PL-12 positive patients have muscle involvement at a frequency of only 52%, but interstitial lung disease at a frequency of 90%.
A number of different autoantibodies are found exclusively in dermatomyositis. For example, antibodies recognizing the chromatin remodeling enzyme Mi-2 are found in as many as 20% of patients with dermatomyositis where they are associated with particularly fulminant initial cutaneous manifestations. However, these patients tend to have a good response to treatment and have a lower risk of associated malignancy. The anti-MDA5 antibodies have been reported only in patients with amyopathic dermatomyositis, especially in those with interstitial lung disease. Anti-small ubiquitin-like modifier 1 (SUMO-1) autoantibodies have also been reported in ˜8% of patients with dermatomyositis. The so-called anti-p155/140 autoantibody is also found exclusively in patients with dermatomyositis; it is associated with an increased risk of malignancy. Unlike other autoantibodies, anti-p155/140 and anti-NXP-2 may also be found in children with dermatomyositis. Children with anti-p155/140 have especially severe skin disease and patients with anti-NXP-2 have an increased risk of developing subcutaneous calcium deposits. However, there was no increased risk of tumors in children with or without these autoantibodies.
Two different myositis-specific antibodies are particularly associated with immune-mediated necrotizing myopathy. Anti-SRP autoantibodies are found in ˜5% of myositis patients and are associated with muscle biopsies showing predominantly myofiber degeneration and necrosis with minimal inflammation. These patients tend to have a rapidly progressive myopathy associated with very high CK levels, early muscle atrophy, dysphagia and, frequently, an incomplete response to immunosuppressive therapy. Patients with anti-HMGCR antibodies are found in ˜5% of myositis patients and have a strong association with a necrotizing muscle biopsy and prior statin exposure; in patients over the age of 50 with these antibodies, 92% reported a prior statin exposure. In such patients, the myalgias and weakness that began during statin therapy persists and progresses for months or years after the statin is withdrawn. Significant improvement (and sometimes resolution) of the myopathy requires immunosuppressive therapy.
Antinuclear antibodies are found in more than half of patients with polymyositis or dermatomyositis and are associated with the presence of antibodies recognizing Mi-2 (a nuclear protein) or an autoantigen associated with one of the other connective tissue diseases. Examples include anti-PM-Scl and anti-Scl-70, autoantibodies found in patients with scleroderma-myositis overlap, and anti-RNP, which is found in patients with mixed connective tissue disease. The erythrocyte sedimentation rate and C-reactive protein are only markedly elevated in about 20% of myositis patients and are not particularly diagnostically useful.
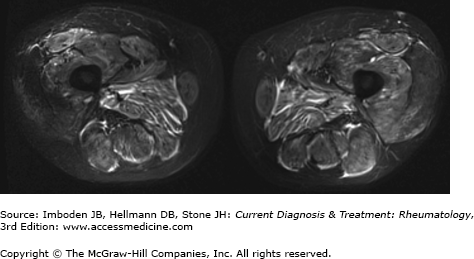
Although neither conventional radiography nor radionuclide imaging have proved particularly useful in patients with muscle diseases, computer-based image analysis using ultrasonography, CT, and MRI can aid in diagnosis and assessment of disease activity. Of these, MRI with T2-weighted images and fat suppression or short tau inversion recovery (STIR) offers the best imaging of soft tissue and muscle (Figure 27–3). MRI plays an important role in the evaluation of management of autoimmune myopathy because of its ability to evaluate muscle edema (as an indicator of active inflammation) and fatty infiltration (as an indicator of chronic disease). MRI can detect early or subtle disease changes as well as patchy muscle involvement. Because of these capacities and the fact that it is noninvasive, MRI may prove superior to EMG in determining the site for muscle biopsy. Furthermore, MRI can be used to assess degree of fascial involvement as well as to semi-quantitatively grade muscle involvement and, therefore, can be used to monitor the response to therapy. This may be particularly useful when trying to differentiate between active myositis and glucocorticoid-induced myopathy. In such situations, the presence of edema in the muscle tissue is indicative of an ongoing inflammatory process. Use of MRI to guide biopsy may lower the rates of false-negative muscle biopsies, which range from 10% to 25%. Repeated MRIs can be used to monitor disease progression and response to therapy. However, treatment with immunosuppressive medications can result in decreased signal intensity on MRI, but histologically detected inflammation may not change significantly.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


