Chapter 3. Connective tissue inflammation, repair and remodelling
CHAPTER CONTENTS
SUMMARY
This chapter examines the different phases of healing and explores the process of scar tissue formation and its implication in restoring or preventing pain-free function. General principles of treatment are applied to each phase, aiming to facilitate healing, and the factors which promote or impede healing are considered.
The different phases of connective tissue healing are not separate and each is overlapped by the other, with one response signalling another until the wound is bridged by scar tissue (Fig. 3.1). After an initial, relatively short bleeding phase, the inflammatory phase prepares the area for healing, the repair phase rebuilds the structure and the remodelling phase provides the final form of the tissue (Hardy 1989, Broughton et al 2006, Watson 2009).
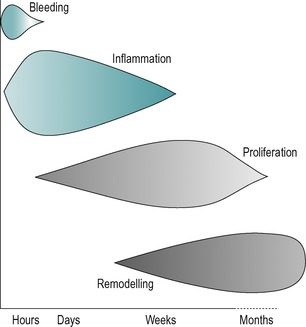 |
| Figure 3.1 Tissue repair phases and timescale. (Online. Available: http://www.electrotherapy.org. Reprinted by permission of Professor Tim Watson.) |
The degree of inflammation in response to injury depends on the degree of trauma. A minor injury causes a minimal response whereas a major injury will produce a significant inflammatory response which will pass through acute, subacute and chronic phases.
Lesions encountered in orthopaedic medicine include acute and chronic muscle belly lesions, ligamentous lesions, overuse tendinopathy, tenosynovitis, arthritis, bursitis and mechanical joint displacements.
Acute inflammation is significant and the patient can usually recall the precise time and mode of onset. Following injury the inflammatory response is rapid with noticeable pain and swelling, which can last for hours or days. With chronic inflammation, the patient cannot usually recollect the onset and the reaction is low-grade with less noticeable pain and swelling. Chronic inflammation may occur as a progression from acute inflammation or as a result of overuse, and can last for weeks, months or even years.
As mentioned in Chapter 2, the inflammatory model in chronic tendon lesions has been challenged and the pathological process may involve degenerative rather than inflammatory changes (Cook et al 2000, Khan & Cook 2000). A short discussion on the aetiology and pathology associated with chronic tendon lesions follows here before the phases of healing are described.
The degenerative changes in chronic tendon lesions include collagen fibre breakdown, increased ground substance, neovascularization, increased number of nerve filaments and increased immunoreactivity of substance P and calcitonin; there are no signs of chemical inflammation in chronically painful tendons (Khan & Cook 2000,Cook et al 2000, Wang et al 2006).
Alfredson et al (2002) concur that pathology is linked to degradation of collagen and hypercellularity. Tendons experiencing tendinosis contain no inflammatory cells but exhibit changes in the collagen fibre ultrastructure (Alfredson et al 2002, Tasto et al 2003, Richards et al 2005) with irregular fibre arrangement and a high concentration of glycosaminoglycans (Alfredson et al 2002). Local hypoxia, repetitive microtrauma or impaired wound healing may also contribute to tendinopathy (Richards et al 2005).
Tendinopathy has been adopted as a more appropriate term since it does not commit to pathology. The terms ‘tendinosis’, ‘paratendinitis’ and ‘tendinitis’ are reserved as histopathological labels that need to be confirmed by histopathological studies.
Tenosynovitis is distinct from tendinopathy; it occurs in ensheathed tendons and involves the synovium rather than the tendon itself. Inflammation of the synovium can produce adhesions between the two layers of the sheath which may produce a palpable crepitus (Cyriax 1982).
Characteristics of tendinopathy include a combination of pain, swelling and impaired performance.
The aetiology of tendinopathy appears to be multifactorial and the pathogenesis is unclear. It has increased in the general population and is found in both those who participate in recreational sport and those with a more sedentary lifestyle, as well as in more elite athletes. Other contributing factors will be discussed for specific tendon lesions, as well as their management, in the relevant chapters that follow.
Repetitive strain can reduce the ability of the tendon to endure further tension, disrupting its microscopic and macroscopic structure, but Rees et al (2006) propose that underuse could also be a factor. Inflammation (i.e. tendinitis or peritendinitis) may be the initial finding in tendon overuse and if this progresses the ensuing focal degeneration (i.e. tendinosis) can lead to partial and complete tears (Józsa & Kannus 1997). Other factors such as vascular supply, age and genetics can play a part in the pathogenesis of tendinopathy (Wang et al 2006). More research is needed to understand the mechanisms of tendinopathy at the tissue, cellular and molecular levels to be able to develop more effective evidence-based treatment protocols for tendinopathy.
Where the pain comes from in tendon lesions is also still unclear and Rees et al (2006) put forward mechanical, vascular and neural theories, suggesting that the pain arises from a combination of factors. Alfredson et al (2002) had suggested previously that neovascularization might be involved.
Structural damage noted in tendinopathy may include partial tearing of the collagen fibres, and an element of rest, especially from the aggravating activity, is appropriate to allow time for healing. The low metabolic activity in tendon causes an extended healing period, (Wang et al 2006). A controlled, graded programme of strengthening, stretching and eccentric exercises can be introduced guided by pain and function (Brukner & Khan 2007); ballistic stretching also has an important role in rehabilitation (Witvrouw et al 2007).
INFLAMMATORY PHASE
The initial inflammatory reaction involves vascular and cellular changes. Injury is rapidly followed by transient vasoconstriction, lasting for 5–10 min, and is succeeded by vasodilatation which may result in haemorrhage. If the lesion is still bleeding at the time of treatment this will necessitate careful management.
Figure 3.2 illustrates the components of the acute and chronic inflammatory responses, which will be referred to below. As described in Chapter 2, connective tissue comprises cells embedded in an extracellular matrix of fibres, with an interfibrillar amorphous ground substance that is assumed within the background spaces in the diagram.
 |
| Figure 3.2 The components of acute and chronic inflammatory process, circulating cells and proteins, cells of blood vessels, and cells and proteins of the extracellular matrix. From Pathologic Basis of Disease 7th edn by V Kumar, N Fausto and A K Abbas. Reprinted by permission of Elsevier Ltd. |
In the early stage of inflammation the blood vessel walls become more permeable and plasma and leukocytes leak into the surrounding tissues as inflammatory exudate or oedema. Swelling may take a few hours to develop and the amount of swelling is determined by the type of tissue involved in the injury. For example, muscle bellies may produce considerable swelling and bleeding but the structure of tendons prevents the collection of fluid and they do not easily swell. Similarly, ligaments themselves do not usually show dramatic swelling but capsular ligaments (e.g. the medial collateral ligament of the knee) may provoke a traumatic arthritis of the joint causing considerable pain and swelling. Swelling may also be restricted physically by fascial bands and intermuscular septa.
The vascular response is directly due to damage of blood vessel walls and indirectly due to the influence of chemical mediators. These chemical mediators include heparin and histamine, released by the mast cells (Fig. 3.2), bradykinin originating from plasma and plasma proteins, serotonin from platelets and prostaglandins, which are hormone-like compounds, produced by all cells in the body.
Heparin temporarily prevents coagulation of the excess tissue fluid and blood in the area. Bradykinins have multiple effects. They are potent mediators of the inflammatory response, can directly cause pain and vasodilatation, can activate the production of substance P and can enhance prostaglandin release. Substance P promotes vasodilatation, increases vascular permeability and stimulates phagocytosis and mast cell degranulation, with the subsequent release of histamine and serotonin. Prostaglandins may provoke or inhibit the inflammatory response. Histamine and serotonin produce a short-lived vascular effect whereas both bradykinins and prostaglandins promote more long-term vasodilatation (Broughton et al 2006). The overall vascular activity is responsible for the gross signs of inflammation: heat ( calor), redness ( rubor), swelling ( tumor), pain and tenderness ( dolor) and disturbed function ( functio laesa) (Peery & Miller 1971).
Inflammation causes pain and tenderness. Mechanical pain is due to mechanical stress, tissue damage, muscle spasm and the accumulated oedema causing excess pressure on surrounding tissues. Chemical pain arises through chemosensitive nerve receptors which are sensitive to histamine, serotonin, bradykinins and prostaglandins which are released into the tissues during this inflammatory phase. However, most nociceptors (pain receptors) are sensitive to more than one type of stimulus. The inflammatory substances may cause extreme stimulation of nerve fibres without necessarily causing them damage.
The nociceptors become progressively more sensitive the longer the pain stimulus is maintained and pro-inflammatory prostaglandins are believed to sensitize nociceptors leading to a state of hyperalgesia, an increased response to a painful stimulus (Wilkerson 1985, Kloth & Miller 1990).
If the tissue is still haemorrhaging, attempts must be made to stop this as blood is a strong irritant and will cause chemical and mechanical pain, as well as prolonging the inflammatory process (Dingman 1973, Evans 1980). This is particularly true of a haemarthrosis where aspiration should be considered.
In the first few hours of the early inflammatory phase fibronectin, a structural glycoprotein which acts as a tissue ‘glue’, appears in the wound, deposited along strands of fibrin in the clot (Broughton et al 2006, Standring 2009). This fibrin–fibronectin meshwork is associated with immature fibroblasts, which are thought to deposit type III collagen fibres to provide a scaffold for platelet adhesion and anchorage for further invading fibroblasts (Nimni 1980, Lehto et al 1985).
In minor injury, the inflammatory process is short and the scar tissue produced is minimal. The red blood cells break down into cellular debris and haemoglobin pigment, and the platelets (Fig. 3.2) release thrombin, an enzyme which changes fibrinogen into fibrin. The fibrin forms a meshwork of fibres which trap the blood clot and an early scar is formed.
If the injury is significant, the next stage of the inflammatory phase is phagocytic. Circulating monocytes modulate into macrophages (Fowler 1989) (Fig. 3.2) and join the resident macrophage population to clear the debris from the site of injury through phagocytic action (Leibovich & Ross 1974). The macrophages increase in great numbers during the first 3 or 4 days (Dingman 1973). They engulf any matter with which they come into contact, clearing the wound environment and preparing it for subsequent repair. As well as performing a phagocytic role, the macrophage acts as a director cell, directing the repair process by chemically influencing an appropriate number of fibroblast cells activated in the area.
Macrophages also have a role in muscle regeneration, stimulating the production of satellite cells that align themselves to muscle fibres where they are capable of forming completely new muscle fibres or restoring damaged muscle fibres (Grefte et al 2007). Further support for their importance is provided by Shen et al (2008) who observed that a reduced number of macrophages in muscle tissue is associated with reduced muscle regeneration. Fisher & Rathgaber (2006) observed muscle regeneration after acute blunt trauma to the gastrocnemius muscle in rats and noted an initial degenerative process with early peaking of macrophages and fibroblasts in the first 3 days, following which numbers of macrophages declined and satellite cells were noted beneath the basal lamina of muscle fibres. A regenerative process then began and after 6 days numerous irregularly arranged sarcomeres were observed, composed of thick and thin microfilaments and Z bands.
A stage of neovascularization is also reached, with capillaries starting to develop after about 12 h and continuing to develop for a further 2 or 3 days (Daly 1990). The new vessels supply oxygen and nourishment to the injured tissues, but are delicate and easily disrupted. Relative immobilization is necessary at this stage and heat is contraindicated as it will cause increased bleeding from the fragile vessels (Hardy 1989).
Inflammation is a normal response to either trauma or infection and to have no inflammatory response would mean that healing would not occur. Too little inflammation will delay healing and too much inflammation will lead to excessive scarring. The so-called PRICE principle of employing protection, relative rest, ice, compression and elevation in the early stages can help to control heat, swelling and pain and to reduce, but not abolish, inflammation.
The control of swelling is important towards regaining function since the greater the amount of inflammatory exudate, the more fibrin will be found in the area which becomes organized into scar tissue (Evans 1980). Although pain inhibits normal movement, graded mobilization should be encouraged as early as possible to promote healing and to avert adverse scar tissue formation.
Pain itself can be reduced further by analgesic drugs. Non-steroidal anti-inflammatory drugs (NSAIDs) modify the inflammatory response, reduce chemical pain and reduce temperature by inhibiting the production of prostaglandins. There are exponents of the so-called ‘NICE’ principle, adding in the prescription of non-steroidal drugs to the usual first-line after-care of injury. However, a counter view is that these should not be prescribed for the first 2 or 3 days after injury since they will tend to delay healing (Boruta et al 1990, Watson 2009). It may be more appropriate to allow the body’s natural inflammatory response to proceed with analgesics such as paracetamol and/or physical measures such as mobilization, massage and electrotherapy as appropriate methods of pain control.
Corticosteroids are contraindicated in the acute inflammatory phase as they inhibit macrophage activity which delays debridement of the wound and scar tissue production by delaying the onset and proliferation of the fibroblasts (Leibovich & Ross 1974, Hardy 1989). Each stage of the inflammatory phase is essential to the repair process and suppression in the early stages will delay healing.
Gentle transverse frictions and controlled mobilization can be initiated early to allow healing in the presence of movement, as guided by the irritability of the tissues. Mobilization is initiated very gently, to start moving the injured connective tissue towards regaining painless function, but without unduly stressing the healing breach. Overstressing the healing tissue would disrupt the early fragile scar and set up a secondary inflammatory response, leading to excess scar tissue formation. Mobilization also has an effect on the mechanoreceptors which is thought to reduce pain.
Gentle frictions together with gentle mobilization will agitate tissue fluid and increase the chance contact of the macrophage with cellular debris, so promoting healing (Evans 1980).
REPAIR PHASE
The repair phase is simultaneous with the inflammatory phase and overlaps the remodelling phase.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


