Combined Immunodeficiency Diseases
Rebecca H. Buckley
Genetic defects in T-cell function lead to an increased susceptibility to infections or to other clinical problems that are more serious than in those disorders characterized by antibody deficiency alone. Those affected usually present during infancy with either common or opportunistic infections; they rarely survive beyond infancy or childhood. Except for the DiGeorge anomaly, no isolated defects of T-cell development occur. B cells require T-cell help for antibody production; therefore, a T-cell defect must of necessity also result in B-cell deficiency, even when the B cells themselves are completely competent. The spectrum of combined immunodeficiency ranges from the syndrome of severe combined immunodeficiency (SCID), in which T-cell function is absent, to those with combined immunodeficiency disorders (CID) in which some T-cell function is present, but not an adequate amount for a normal life span. Recent discoveries of the molecular causes of many of these defects have led to a new understanding of the flawed biology underlying this ever-growing number of defects (Table 431.1). This chapter reviews the current information on some, but by no means all, of these defects.
DIGEORGE SYNDROME
Pathophysiology and Comorbidity
Thymic hypoplasia results from dysmorphogenesis of the third and fourth pharyngeal pouches during early embryogenesis; this leads to hypoplasia or aplasia of the thymus and parathyroid glands. Other structures forming at the same age also are affected frequently, resulting in anomalies of the great vessels (right-sided aortic arch), esophageal atresia, bifid uvula, upper limb malformations, congenital heart disease (conotruncal, atrial, and ventricular septal defects), a short philtrum of the upper lip, hypertelorism, an anti-mongoloid slant to the eyes, mandibular hypoplasia, and low-set, often notched ears. A variable degree of hypoplasia of the thymus and parathyroid glands (partial DiGeorge syndrome) is more frequent than total aplasia. Those with complete DiGeorge syndrome are highly susceptible to infections by opportunistic pathogens and to graft-versus-host disease (GVHD) from nonirradiated blood transfusions.
DiGeorge syndrome has occurred in both males and females; rare cases of autosomal dominant inheritance have been reported. Microdeletions of specific DNA sequences from chromosome 22q11.2 (the DiGeorge chromosomal region) have been shown in a majority of, but certainly not all, patients with the clinical features of this syndrome. There appears to be an excess of 22q11.2 deletions of maternal origin. Another deletion associated with DiGeorge and velocardiofacial syndromes is on chromosome 10p13.
Clinical Manifestations and Complications
Patients with DiGeorge syndrome may be mildly to severely lymphopenic, due to the fact that the decrease in the number of circulating CD3+ T cells depends on the degree of thymic deficiency. Immunoglobulin concentrations are usually normal, although sometimes IgE is elevated and IgA may be low. Responses of blood lymphocytes after mitogen stimulation have been absent, reduced, or normal. Thymic tissue, when found in the partial form, contains Hassall’s corpuscles and a normal density of thymocytes; corticomedullary distinction is present.
Lymphoid follicles usually are present, but lymph-node paracortical areas and thymus-dependent regions of the spleen show variable degrees of depletion.
Lymphoid follicles usually are present, but lymph-node paracortical areas and thymus-dependent regions of the spleen show variable degrees of depletion.
TABLE 431.1. LOCATIONS OF FAULTY GENES IN CELLULAR IMMUNODEFICIENCY DISORDERS | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
Therapy
Treatment of the complete form of DiGeorge syndrome should begin as soon as possible after diagnosis. Transplantation of cultured, mature thymic epithelial explants has successfully reconstituted the immune function of several infants with the complete DiGeorge syndrome. A few patients with complete DiGeorge syndrome have developed T-cell function after human leukocyte antigen (HLA)-identical bone marrow transplantation by adoptive transfer of donor T cells, but this treatment cannot correct the thymic defect. No immunologic treatment is needed for the partial form. If patients with the partial DiGeorge syndrome do not have a severe cardiac lesion, they have few clinical problems except that some experience seizures and developmental delay.
SEVERE COMBINED IMMUNODEFICIENCY
Pathophysiology
Severe combined immunodeficiency, or SCID, is a fatal syndrome of diverse genetic cause characterized by profound deficiencies of T- and B-cell (and sometimes natural killer [NK] cell) function. These infants also lack the ability to reject foreign tissue and are, therefore, at risk for GVHD from maternal T cells that cross into the fetal circulation while the SCID infant is in utero, or from T lymphocytes in nonirradiated blood products (Fig. 431.1) or allogeneic bone marrow.
In the five decades since the initial description of SCID, it has become evident that the genetic origins of this condition are extremely diverse (Fig. 431.2). X-linked SCID (SCID-X1) is the most common form, accounting for approximately 46% of cases in the United States (Fig. 431.2). In addition, mutated genes on autosomal chromosomes have been identified in nine genetic types of SCID: adenosine deaminase (ADA) deficiency, Janus kinase 3 (Jak3) deficiency, interleukin-7 receptor alpha-chain deficiency (IL-7R), recombinase-activating gene (RAG-1 or RAG-2) deficiencies, Artemis deficiency, CD45 deficiency, and CD3 delta (CD3δ) chain and epsilon (CD3ε) deficiencies; and there are likely other causes yet to be discovered (Table 431.2).
Clinical Manifestations and Complications
Affected infants present within the first few months of life with frequent episodes of diarrhea, pneumonia, otitis, sepsis, and cutaneous infections. Persistent infections with opportunistic organisms such as Candida albicans, Pneumocystis jiroveci,
varicella-zoster virus (Fig. 431.3), parainfluenza 3 virus, respiratory syncytial virus, adenovirus, cytomegalovirus, Epstein-Barr virus, and bacillus Calmette-Guérin lead to death. Infants with SCID are lymphopenic due to the fact that they lack T cells. They have an absence of lymphocyte proliferative responses to mitogens, antigens, and allogeneic cells in vitro, even on blood samples collected in utero or from the cord blood.
varicella-zoster virus (Fig. 431.3), parainfluenza 3 virus, respiratory syncytial virus, adenovirus, cytomegalovirus, Epstein-Barr virus, and bacillus Calmette-Guérin lead to death. Infants with SCID are lymphopenic due to the fact that they lack T cells. They have an absence of lymphocyte proliferative responses to mitogens, antigens, and allogeneic cells in vitro, even on blood samples collected in utero or from the cord blood.
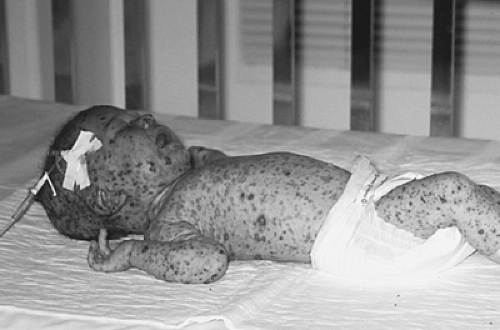 FIGURE 431.1. An infant with SCID who developed lethal graft-versus-host disease from a non-irradiated packed red blood cell transfusion. (Reproduced with permission from Buckley RH. A historical review of bone marrow transplantation for immunodeficiencies. JACI 2004;113:792.) |
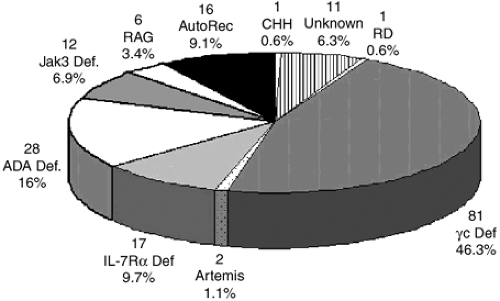 FIGURE 431.2. Relative frequencies of the different genetic types of SCID among 175 patients seen consecutively by the author. |
Diagnosis
Physicians caring for newborns must be aware of the normal range for the cord blood absolute lymphocyte count (2,000 to 11,000/mm3) and arrange for T-cell phenotypic and functional studies to be performed on blood from neonates with values below 2,500/mm3. Unfortunately, most SCIDs are not diagnosed until 6 to 7 months of age, when they present with serious infections. At that age, the normal absolute lymphocyte count is much higher, with the mean being 7,500/mm3 and the lower limit of normal 4,000/mm3. Serum immunoglobulin concentrations are diminished to absent, and no antibody formation occurs after immunization. Typically, all SCID patients have very small thymuses (usually less than 1 g), which fail to descend from the neck, contain no thymocytes, and lack corticomedullary distinction and Hassall’s corpuscles. However, the thymic epithelium is normal, and the results of bone marrow stem cell transplantation have shown that these tiny thymuses are capable of supporting the maturation of stem cells into normal T cells. Thymus-dependent areas of the spleen are depleted of lymphocytes in SCID patients, and lymph nodes, tonsils, adenoids, and Peyer’s patches are absent or extremely underdeveloped.
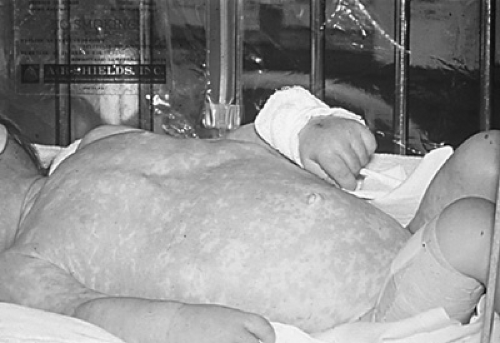 FIGURE 431.3. Infant with X-linked SCID who experienced varicella three times before the diagnosis of SCID was made. The child survived because of the availability of acyclovir and the ability to give a haploidentical bone marrow transplant from his mother. (Reproduced with permission from Buckley RH. A historical review of bone marrow transplantation for immunodeficiencies. JACI 2004;113:792.) |
TABLE 431.2. TEN ABNORMAL GENES IN SCID | |
|---|---|
|
AUTOSOMAL RECESSIVE SEVERE COMBINED IMMUNODEFICIENCY DISEASE CAUSED BY ADENOSINE DEAMINASE DEFICIENCY
Pathophysiology
An absence of the enzyme adenosine deaminase (ADA) was the first discovered molecular cause of SCID. It was first reported in 1972, and has since been observed in approximately 16% of patients with SCID, being the second most common cause of SCID (Table 431.2, Fig. 431.2). The gene encoding ADA is on chromosome 20q13-ter.
Clinical Manifestations and Complications
Certain distinguishing features of ADA deficiency can be noted, including the presence of multiple skeletal abnormalities of chondroosseous dysplasia on radiographic examination; these occur predominantly at the costochondral junctions, at the apophyses of the iliac bones, and in the vertebral bodies (causing a “bone-in-bone” effect). ADA deficiency results in pronounced accumulations of adenosine, 2′-deoxyadenosine, and 2′-O-methyladenosine. These toxic metabolites directly or indirectly lead to apoptosis of thymocytes and circulating lymphocytes, which causes the immunodeficiency. ADA-deficient infants usually have a much more profound lymphopenia than those with other types of SCID, with mean absolute lymphocyte counts of less than 500/mm3. They thus have a T-B-NK-lymphocyte phenotype (Table 431.3).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


