This article addresses the pathophysiology, diagnostic approaches, and therapeutic options in the more common forms of muscular dystrophy, especially those seen in pediatric and young adult populations. The major emphasis is on the dystrophinopathies because their treatment options are templates for those used in various other forms of dystrophy. Most patients with cardiomyopathy are treated with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, with other agents added as the disease progresses. Destination therapies and transplantation options are mentioned where appropriate. Some dystrophies can have significant conduction abnormalities requiring pacemaker treatment. Others with ventricular tachydysrhythmias may necessitate internal cardiac defibrillator placement.
- •
Various cardiac manifestations are seen in different types of muscular dystrophy, including cardiomyopathy and disturbances of conduction.
- •
Treatment strategies are evolving, with afterload reduction largely replacing use of digoxin in patients with cardiomyopathy with decreased ejection fraction.
- •
Careful genetic analysis allows proper classification of the various muscular dystrophies, resulting in proper diagnostic and treatment choices.
Introduction
This article addresses the present concepts regarding treatment of the more common neuromuscular diseases in children and young adults. These evolving strategies will likely be refined over time. Some are surrounded with debate, and differences are discussed. Treatment is predicated on accurate diagnosis, particularly genetic diagnosis, which is detailed in other articles in this issue. Cardiac diagnostic methods are briefly discussed. For more detail, the reader is referred to the chapter entitled “The Heart in the Muscular Dystrophies” in Moss and Adams’ Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adult .
Cardiac diagnostic methods
Common methods for cardiac evaluation include electrocardiography, Holter monitoring, Doppler echocardiography, and MRI. These modalities are discussed as they apply to the various neuromuscular diseases.
Electrocardiography
The electrocardiogram shows heart rhythm, rate, axis in the frontal plane, and changes in voltages interpreted as patterns of hypertrophy.
Holter Monitoring
Holter monitoring shows the heart rate patterns over a 24- or 48-hour time span. It can also reveal the presence of arrhythmias and ST-T wave abnormalities.
Cardiac Ultrasound
Doppler echocardiography shows cardiac anatomy and patterns of blood flow, which is especially important for evaluating left ventricular (LV) function. Areas of akinesis (lack of motion) or dyskinesis (abnormal motion) can be seen. LV remodeling, or rounding of the diseased ventricle, found with cardiomyopathy can be measured through comparing LV diastolic dimension and LV length (sphericity index). Systolic function can be measured through shortening fraction (LV end-diastolic dimension-LV end-systolic dimension vs LV end-diastolic dimension) and ejection fraction (LV systolic area vs LV diastolic area) ( Figs. 1 and 2 ). Diastolic function can be assessed through measuring the LV diastolic dimension, relationship of mitral inflow velocities (E wave/A wave ratio), and tissue Doppler. Systolic and diastolic time intervals also reflect cardiomyopathy. The presence of aortic regurgitation and/or mitral regurgitation also can reflect LV dysfunction. Tricuspid and pulmonary regurgitation jet quantification can be used to predict pulmonary arterial pressures. With experience and perseverance, adequate imaging can be obtained in most patients with muscular dystrophy.
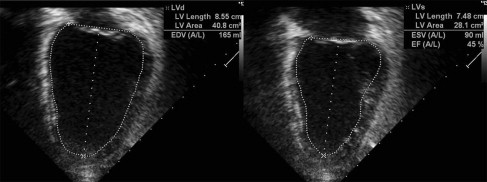
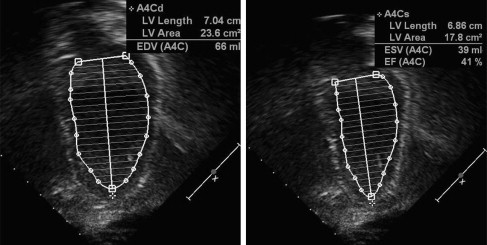
MRI
MRI allows superior anatomic definition and functional analysis compared with echocardiography but is more expensive and can cause claustrophobic reactions. Patients younger than 10 years usually require anesthesia. Scheduling MRI testing can be a problem because of limitations in availability. Furthermore, the presence of metallic objects, such as pacemakers or rods used in scoliosis surgery, can interfere with imaging.
Cardiac diagnostic methods
Common methods for cardiac evaluation include electrocardiography, Holter monitoring, Doppler echocardiography, and MRI. These modalities are discussed as they apply to the various neuromuscular diseases.
Electrocardiography
The electrocardiogram shows heart rhythm, rate, axis in the frontal plane, and changes in voltages interpreted as patterns of hypertrophy.
Holter Monitoring
Holter monitoring shows the heart rate patterns over a 24- or 48-hour time span. It can also reveal the presence of arrhythmias and ST-T wave abnormalities.
Cardiac Ultrasound
Doppler echocardiography shows cardiac anatomy and patterns of blood flow, which is especially important for evaluating left ventricular (LV) function. Areas of akinesis (lack of motion) or dyskinesis (abnormal motion) can be seen. LV remodeling, or rounding of the diseased ventricle, found with cardiomyopathy can be measured through comparing LV diastolic dimension and LV length (sphericity index). Systolic function can be measured through shortening fraction (LV end-diastolic dimension-LV end-systolic dimension vs LV end-diastolic dimension) and ejection fraction (LV systolic area vs LV diastolic area) ( Figs. 1 and 2 ). Diastolic function can be assessed through measuring the LV diastolic dimension, relationship of mitral inflow velocities (E wave/A wave ratio), and tissue Doppler. Systolic and diastolic time intervals also reflect cardiomyopathy. The presence of aortic regurgitation and/or mitral regurgitation also can reflect LV dysfunction. Tricuspid and pulmonary regurgitation jet quantification can be used to predict pulmonary arterial pressures. With experience and perseverance, adequate imaging can be obtained in most patients with muscular dystrophy.
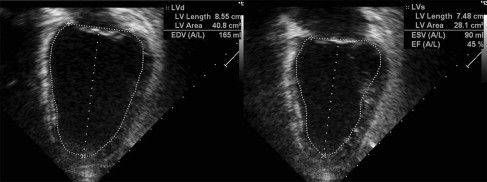
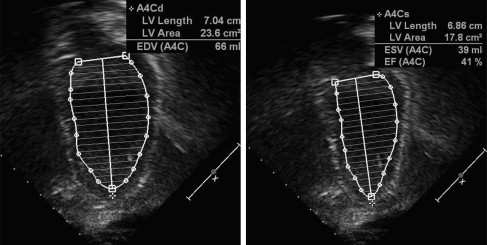
MRI
MRI allows superior anatomic definition and functional analysis compared with echocardiography but is more expensive and can cause claustrophobic reactions. Patients younger than 10 years usually require anesthesia. Scheduling MRI testing can be a problem because of limitations in availability. Furthermore, the presence of metallic objects, such as pacemakers or rods used in scoliosis surgery, can interfere with imaging.
Muscular dystrophies involving the heart
Dystrophinopathies
The 2 common dystrophinopathies are Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy. According to clinical definitions of these X-linked disorders, patients who are ambulatory at 15 years of age have Becker muscular dystrophy. The genetic definition separates the 2 based on the DMD gene exon deletion: out-of-frame for Duchenne and in-frame for Becker. Furthermore, on muscle biopsy, little or no (<5%) dystrophin is seen in patients with DMD, whereas more is present in patients with Becker muscular dystrophy. Symptoms of heart failure are virtually absent in wheelchair-dependent patients with DMD, but ambulatory patients with Becker muscular dystrophy will have symptoms of shortness of breath and fatigue with ambulation.
DMD
History and physical examination
Patients with DMD are rarely symptomatic unless they are ambulatory. An apical S3 and S4 may be present in advanced cardiomyopathy. Additionally, a mitral regurgitation murmur may be audible. Patients with advanced disease may have signs of heart failure, including neck vein distention, sacral edema, peripheral edema, and hepatomegaly.
Pathology
Cardiac involvement in DMD is caused by fibrosis and scarring that proceeds from the epicardium to the endocardium, starting generally at the region behind the posterior mitral valve apparatus. This scarring spreads downward progressively toward the apex and around the heart, ultimately leading to cardiomyopathy. The myopathy is not always dilated, because the scarred areas do not have expansile properties. This process has been confirmed in vivo by MRI studies. A similar process is seen in Becker muscular dystrophy, and a recent study has shown that determining the exon deletion site has predictive value in estimating the timing of cardiomyopathy onset.
Electrocardiography
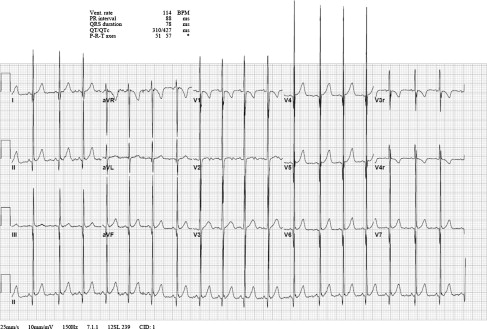
The classic findings in DMD include a shortened PR interval, right ventricular hypertrophy (RVH), and Q waves in the inferolateral leads. Previous literature suggested that Q waves in leads I and aVL were common and that RVH was proportional to the degree of myopathy, suggesting that LV fibrosis interfered with LV forces. A large study showed that findings are present in approximately half of patients (43% short PR, 37% RVH, 34% Q wave in V5, V6) ( Fig. 3 ), with no relationship to the degree or presence of cardiomyopathy as defined by an echocardiography-derived ejection fraction of less than 55%. Furthermore, the presence of Q waves was more often seen in the inferolateral leads than the anterior leads. An unpublished serial evaluation of R wave amplitudes in V1 showed no significant progression of that waveform over time in patients with cardiomyopathy of DMD (Thrush PT, Allen HD, unpublished data, 2012).
Holter monitoring
Several studies have shown abnormally fast average heart rates in patients with DMD, regardless of the presence of cardiomyopathy. This condition is termed disordered automaticity , which is generally defined as an average heart rate exceeding 100 beats per minute in patients older than 12 years. It is attributed to a sympathetic imbalance. Older boys (and some younger ones) can have atrial and ventricular ectopy, heart block, and either atrial or ventricular tachydysrhythmias.
Doppler echocardiography studies
Anatomically, LV posterior wall thinning can occasionally be appreciated, along with contractile and relaxation abnormalities. Using the parameters mentioned earlier, as the disease advances, areas of akinesis or dyskinesis can be seen, accompanied by some with LV dilation, decreased shortening fraction, rounding (remodeling) shown by a sphericity index approaching unity, and decreased ejection fraction. Diastolic abnormalities can be seen with increasing mitral valve inflow Doppler E and A wave ratios and lateral and medial tissue Doppler ratios approaching unity.
MRI studies
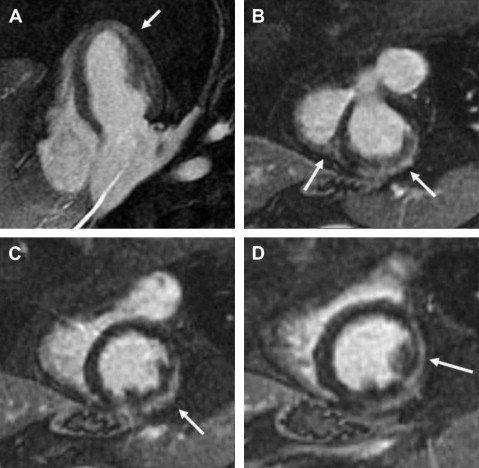
MRI can show the presence and distribution of myocardial fibrosis and offer a measurement of contractility and circumferential strain ( Fig. 4 ). These findings precede the cardiomyopathy defined by an ejection fraction of less than 55% shown on echocardiographic studies. Careful analysis of ejection fraction is also possible with MRI.
Laboratory analysis: biomarkers
Serum brain natriuretic peptide (BNP) and α-atrial natriuretic peptide (α-ANP) levels are elevated in the presence of LV dysfunction in adults and in some boys with DMD and advanced heart failure.
Treatment of Cardiomyopathy in Dystrophinopathy
Timing of treatment initiation
Ideally, boys with DMD should be referred to a cardiologist for an initial evaluation at the time of diagnosis. Changes in the heart have been documented from an early age, with 53% of children aged 5 years and younger showing an abnormality on electrocardiogram. Follow-up surveillance should continue every other year until the child begins to have symptoms or reaches the age of 10 years, when yearly follow-up should be initiated.
When to begin treating these patients with pharmacologic management is debated. Because these boys may not have symptoms, the standard has been to commence therapy if the echocardiography-derived ejection fraction is less than 55%. This timing is parallel with that of treatment of other forms of cardiomyopathy. Some have initiated treatment if, in addition to the presence of echocardiographic abnormalities, biomarkers (α-ANP, BNP) are abnormal.
Preventive treatment
Some centers advocate beginning treatment before echocardiographic abnormalities manifest, arguing that all of these boys have cardiomyopathy (raising the question, what is a cardiomyopathy?) regardless of the later-stage echocardiographic abnormalities. Duboc and colleagues conducted a trial of placebo versus angiotensin-converting enzyme inhibitor (ACE-I) in boys with DMD who had normal ejection fraction on radioisotope analysis, with all boys placed on the ACE-I after 3 years. Although all patients showed progressive deterioration in function, no significant difference was seen between the groups in their ejection fractions over time, regardless of whether they were treated. More patients in the placebo group died than in the treated group at a 10-year follow-up. Because of the increased death rate (Kaplan-Meyer survival lower in the placebo group; P = .13), the investigators concluded that boys should be treated prophylactically after the age of 9.5 years. Although the causes of deaths were not detailed, the study has resulted in many centers adopting a preventive treatment approach.
An important MRI study by Hor and colleagues showed that boys with normal echocardiographic ejection fraction had fibrosis and decreased peak circumferential strain on MRI. Patients younger than 10 years showed abnormal strain, and older boys showed a further decline in strain, with abnormal ejection fraction appearing at a mean age of 15.8 years.
A mouse model of DMD showed a protective effect of Aldactone in combination with an ACE-I on both skeletal and cardiac muscle. This effect may be from the antifibrotic effects of these medications (transforming growth factor β inhibition) in treated mice versus controls. Clinical trials are obviously needed before this may be potentially translatable to boys with DMD.
Drugs and treatments used in cardiomyopathy of DMD
In the past, standard treatment was with digoxin and diuretics; however, because of the arrhythmogenic potential of digoxin, it has largely fallen out of favor.
The standard treatment of cardiomyopathy in adults has been ACE-I, and several centers have used the same concept in boys with cardiomyopathy of DMD. At the authors’ center, an analysis of 42 boys showed that the mean age of onset of cardiomyopathy was 14.1 years, with a range of 7 to 27.3 years. Use of ACE-I resulted in ejection fraction improvement from a mean of 44% to 52% ( P ≤.001). Addition of, or initial use of, a β-blocker before ACE-I initiation (usually for disordered automaticity) did not change the result ( P = .947).
Angiotensin receptor blocker treatment can be effective and is associated with less cough (a rare complication of ACE-I). Furthermore, at least in the mdx mouse, skeletal and cardiac muscle regeneration has been seen. Clinical studies are needed to determine whether this occurs in humans. One such Muscular Dystrophy Association–sponsored study is underway.
Whether eplerenone or aldactone (both aldosterone antagonists) is effective in treating cardiomyopathy or reducing skeletal muscle degeneration requires careful and prospective double-blinded clinical studies.
For severe heart failure, hospitalization and treatment with milrinone may be useful. One study has advocated using intravenous milrinone as home therapy, but this is not without potential adverse events, and most centers reserve this treatment for the inpatient arena.
Idebenone (a benzoquinone with antioxidant properties) improves respiratory chain function and cellular energy production and was found to be beneficial in a trial in patients with DMD. Improvement in pulmonary function was also seen. A larger trial is ongoing. Trials are also underway assessing the use of sildenafil and carvedilol.
Atrial and ventricular arrhythmias are treated as per standard. For disordered automaticity, β-blocker therapy is used. If ventricular tachydysrhythmias are present, sotalol (a class III antiarrhythmic) or flecainide (a class Ic antiarrhythmic) may be used.
Automatic implantable cardioverter defibrillator placement may be helpful in patients with ventricular tachydysrhythmias, and is also considered in patients with an ejection fraction less than 15%, similar to the approach used in adults with severe congestive heart failure.
Another observation has been that boys with DMD can have hyperkalemic or hyperthermic reactions to inhalant anesthetic agents or muscle relaxants, suggesting that these agents should be avoided during general anesthesia.
Use of nocturnal bilevel positive airway pressure (BiPAP) for respiratory support also benefits boys with cardiomyopathy. Although BiPAP results in decreased preload, the primary benefit is secondary to decreased LV transmural pressure and, ultimately, decreased afterload. With advances in pulmonary and cardiac treatment, these patients, although lacking skeletal muscle function, now survive longer.
Some providers consider using destination therapy (eg, extracorporeal membrane oxygenation, acutely, or left ventricular–assist devices, which can be used over a long term) in patients with DMD with severe cardiomyopathy, using the survival argument, although the ethics of this approach can be debated. The same argument can be made for cardiac transplantation. The ultimate hope is that gene therapy may reverse or stop the disease. Whether the cardiac myocyte response to gene therapy will be the same as the skeletal muscle response remains to be seen.
Related posts:
 Neuromuscular Disease Management and Rehabilitation, Part II: Specialty Care and Therapeutics
Nutrition Assessment and Management in Amyotrophic Lateral Sclerosis
Current Pharmacologic Management in Selected Neuromuscular Diseases
Novel Approaches to Corticosteroid Treatment in Duchenne Muscular Dystrophy
Mobility-Assistive Technology in Progressive Neuromuscular Disease
Using Palliative Care in Progressive Neuromuscular Disease to Maximize Quality of Life
Neuromuscular Disease Management and Rehabilitation, Part II: Specialty Care and Therapeutics
Nutrition Assessment and Management in Amyotrophic Lateral Sclerosis
Current Pharmacologic Management in Selected Neuromuscular Diseases
Novel Approaches to Corticosteroid Treatment in Duchenne Muscular Dystrophy
Mobility-Assistive Technology in Progressive Neuromuscular Disease
Using Palliative Care in Progressive Neuromuscular Disease to Maximize Quality of Life
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


