Pre-injury biomarker generation. As illustrated in Fig. 25.1, biomarker generation may begin in the pre-injury phase among individuals who exhibit abnormal biomechanical movement patterns, leading to aberrant mechanical joint loading and biological changes. Cartilage degeneration may occur because of changes in loading patterns. A study from the U.S. Military Academy supports these pre-injury differences in serum biomarker levels [14]. Serum C-terminal propeptide of Type II collagen (CPII), Type I and II collagenase-generated cleavage epitope (C1,2C), and Type II collagenase-generated cleavage epitope (C2C) were compared pre- and post-injury in 45 anterior cruciate ligament (ACL) injured and 45 control participants who were matched on age, sex, height, and weight. Because these biomarkers in subjects of this age can be generated from open growth plate cartilage, it is particularly important, as done here, to match subjects in the injury and non-injury groups by age. Biomarker levels were expected to be comparable at baseline/pre-injury for ACL injured cases and controls, diverging at follow-up/post-injury. However, cases and controls differed not only at follow-up/post-injury but also at baseline/pre-injury, with biomarker levels of serum CPII and C1,2C tending to be higher among those participants who subsequently injured their ACLs compared to those without injury. These differences are consistent with the concept of a pre-injury biomarker risk profile that may be related to cartilage metabolism and ACL injury risk.
Post-injury biomarker generation. The link of injury and osteoarthritis (OA) is well-supported [13, 15–17]. The acute inflammatory response from the injury itself, along with the trauma associated with surgical repair, may contribute to biomarker generation, mainly biomarkers of inflammation, cartilage turnover, and joint metabolism [18]. After the initial injury or surgical repair, changes to the joint tissues, particularly in a weight-bearing joint, may result in a decline in the tissues’ ability to manage dynamic forces, particularly with movements requiring rapid acceleration and deceleration, such as running and jumping. Alterations in movement and joint loading patterns may contribute to an uneven distribution of joint forces during daily activities or sport, with overloading of some cartilage regions and insufficient loading of others [13, 16, 17].
Herein, we summarize current efforts to validate and establish criteria for identifying the trajectory to PTOA early in its course; these would serve as endpoints for biomarker qualification. Secondly, we summarize preclinical and clinical studies that report biomarker data and in keeping with the theme of the book, discuss these from the perspective of the insights they can provide to disease pathogenesis, diagnosis and management. Finally, we discuss what biomarkers tell us about pathogenesis in PTOA compared to idiopathic OA.
Advances in Qualification Endpoints for PTOA Biomarkers
When using a biomarker as a substitute for a clinically meaningful endpoint, one must first be clear about the clinically meaningful endpoint for which the biomarker is a proposed surrogate [19]. The insensitivity of radiographic endpoints for OA has led to ongoing intensive efforts to evaluate other imaging and biochemical biomarkers for their ability to detect worsening of OA more quickly and meaningfully [20]. This same issue has plagued attempts to prevent PTOA. Namely, improvement of the long-term outcomes of joint injury have been hampered by the lack of qualification endpoints for early events that identify an adverse trajectory, i.e., one headed to PTOA 5–20 years later. Recently the Arthritis Foundation has launched an OA Flagship Initiative—the Anterior Cruciate Ligament (ACL) Intervention study (https://www.arthritis.org/research/funded-research/acl-feasibility-trial/) to test the feasibility of multi-site coordination of T1rho and other magnetic resonance imaging in the very acute and subacute setting of an ACL injury. This study could be pivotal to the establishment of a clinical trial paradigm for testing adjunctive early therapeutics, and monitoring with biomarkers from the onset of injury to improve and someday hopefully fully prevent PTOA.
The most useful biochemical biomarkers for diagnosing and monitoring susceptibility to PTOA are macromolecules originating from joint structures whose levels in serum, urine, and synovial fluid reflect processes taking place locally in the joint. The test of the ability of a biomarker to report on local events is best addressed by analyses of synovial fluid and matched serum; an optimal candidate biomarker is likely to have synovial fluid concentrations that are equal to or greater than serum concentrations and serum concentrations that correlate with synovial fluid concentrations. A few biomarkers to date seem to meet these criteria [21] but a great deal more work is needed to expand the armamentarium of systemic biomarkers that could be used to monitor joint health and metabolism pre- and post-injury to the joint.
PTOA Biomarkers in Preclinical Studies
Both in vitro and in vivo PTOA models are useful for defining biomarkers of early OA and for monitoring response to pharmacological and nonpharmacological (including surgery) therapy. In vitro loading of cartilage explants can provide useful insights into the biomarkers generated by injury and a system in which to test the efficacy of interventions designed to prevent the development of PTOA. In one study, the magnitudes of mechanical stress in a range of physiological to hyperphysiologic strains increased the release of cartilage oligomeric matrix protein (COMP) in proportion to the magnitude of dynamic mechanical stress, increased keratan sulfate, chondroitin sulfate and glycosaminoglycan (GAG) in a bimodal pattern, and decreased protein and proteoglycan synthesis at the highest level of stress [22]. In a recent study, a single impact led to detrimental effects on cell viability, and release of GAG and prostaglandin E2 to the media, which were primarily strain dependent [23].
Nearly all animal models of OA are PTOA models. Therefore, close attention to the results of biomarker analyses and interventions in these models could provide major insights for monitoring and prevention of PTOA in humans. A recent study in minipigs demonstrated that upregulation of genes coding for proteins capable of degrading cartilage extracellular matrix occurred within the first few days after anterior cruciate ligament injury; this response was in chondrocytes, cells in the synovium, ligament and scar tissue located between the torn ends of the ligament [24]. Matrix metalloproteinase (MMP)-1 gene expression was upregulated in the articular cartilage, synovium and ligament, MMP-13 expression was suppressed in the articular cartilage, but upregulated 100-fold in the synovium and ligament, and ADAMTS-4 (a disintegrin and metalloproteinase with thrombospondin motifs 4) was upregulated in the synovium and ligament but not in the articular cartilage. They noted that the concentration of collagen degradation fragments (C2C) in the synovial joint fluid nearly doubled in the first 5 days after injury. In the superhealer MRL/MpJ mice compared with non-superhealer mice, protection from PTOA was associated with lower protein levels of IL-1alpha and IL-1beta in the synovial fluid, serum, and joint tissues; higher systemic levels of the anti-inflammatory cytokines IL-4 and IL-10 [25]; lower gene expression of tumor necrosis factor alpha, IL-1beta, macrophage inflammatory proteins and macrophage-derived chemokine (CCL22) in the synovial tissue; and reduced acute and late-stage infiltration of synovial macrophages [26]. The synovial fluid biomarker analyses in mice were made possible by the use of a novel synovial fluid recovery method suitable for very small joints [27]. These data show strong associations of joint tissue inflammation with the development and progression of PTOA in mice. Taken together, these results demonstrate a holistic response of the whole joint unit to injury. They also support the hypothesis that acute events, arising immediately at the time of injury, play a key role in susceptibility to PTOA in the long-term and may need to be appropriately neutralized to prevent PTOA.
A mouse fracture model supports the role of inflammation and cytokines in acute joint injury, showing that acute joint pathology and synovial inflammation are associated with increased intra-articular fracture severity in the mouse knee [28]. This model has provided further support for the hypothesis that immediate and early events after injury play a role in PTOA development [29, 30]; in this regard, a one-time intra-articular delivery of a small amount (0.9 mg) of IL-1Ra immediately after fracture, but not 4 weeks of continuous systemic delivery (1 mg/day), dramatically reduced cartilage degeneration and synovial inflammation [29]. Moreover, intra-articular delivery of purified mesenchymal stem cells in this model system prevented PTOA [31]. Interestingly, stem cell therapy improved OA scores without reducing the degree of synovial hyperplasia after fracture. Because the mesenchymal stem cells were capable of inhibiting the proliferation of in vitro stimulated splenocytes, the authors proposed their mode of action was immunomodulatory.
In an animal model, intra-articular IL-1 superimposed on previous joint injury caused a more rapid and more severe arthritis [32]. IL-1 is released as part of the acute inflammatory response following tissue damage, causing local increases of the proinflammatory cytokines IL-6, tumor necrosis factor alpha (TNF-α), and transforming growth factor beta (TGF-ß1) [33]. TGF-ß1 is a profibrotic cytokine that plays a key role in normal wound healing and in the development of progressive tissue fibrosis [34–36]. These cytokines contribute to inflammation and fibrogenesis by stimulating myofibroblasts, fibroblasts, and extracellular accumulations of collagen and fibronectin [37, 38]. Taken together, these data show that inflammation and biological factors play a key role in PTOA development following injury. A comprehensive understanding of these immediate and early molecular events and their timecourse will go far to establishing a molecular biomarker profiling that could be used to prognosticate, at particular times, the anticipated susceptibility to a PTOA trajectory. Moreover, the growing pharmacopeia of biological agents to inhibit these factors, including for instance the key OA-related cytokine IL-1, provides new avenues for prevention of PTOA.
PTOA Biomarkers in Clinical Studies
A cascade of biomarker changes occurs after injury. This timecourse of biomarker changes has been noted to recapitulate the degradation of matrix components observed in cartilage explants in vitro upon addition of proinflammatory cytokines [18]; namely, changes are preceded by proteoglycan loss followed by collagen degradation, considered an irreversible insult to joint integrity. These observations reveal that the onset of pathological cartilage catabolism is immediate, like a “heart attack of the joint”. This timecourse of joint metabolic disturbances suggests, as mentioned above, that immediate action may be required to alter this course of deleterious events.
After knee injury, cartilage degradation is favored over repair, with increased collagen cleavage [39]. Within the first month after joint injury in humans, Lohmander has documented synovial fluid elevations of cartilage proteoglycan fragments and metalloproteinases [40], collagen fragments [41] and persistent elevations of these molecules over decades [42–46]. Lohmander 1993 [42] showed sustained, 20-year elevations following joint injury, of synovial fluid proteoglycan fragments, MMP-3 (Stromelysin-1), and the ratio of MMP-3/TIMP (tissue inhibitor of metalloproteinases-1). The sustained increased release of cartilage macromolecular fragments is thought to be responsible for the frequent development of PTOA in patients with injuries. Joint instability in addition to the constitutive and excess loss of cartilage macromolecules into synovial fluid after severe joint injury likely contribute to episodic clinical flares and inflammation that further contribute to OA progression [47]. Of note however, the timecourses of the intra-articular cytokine levels appear to vary widely in any given individual [48]; further study of inter-individual variations in response to injury will likely yield insights into risk profiles and mechanisms for effectively neutralizing the trajectory to PTOA.
Figure 25.2 provides a graphic of some potentially useful biomarkers for monitoring susceptibility to PTOA and the impact of interventions. These biomarkers include macrophage-associated cell products (IL-1α, IL-1β, IL-1Ra [receptor antagonist], IL-6, IL-8, IL-10, IL-18, granulocyte-macrophage colony-stimulating factor [GM-CSF], TNFα, and MMP activity); other relevant biomarkers include matrix-associated joint tissue markers: total GAG levels, type II collagen degradation (C2C, urinary C-terminal telopeptide of type II collagen [uCTXII]) and synthesis (CPII) epitopes, COMP, and hyaluronan, and bone related N-terminal telopeptide (NTX-I), to indicate the degree of cartilage degradation and bone turnover in the setting of acute injury [41, 49–56]. In addition, high sensitivity C-reactive protein can be monitored as a marker of inflammation. Total GAG levels correlate with severity of chondral damage in the setting of acute injury [57, 58]. GAG is also released from cartilage in the presence of inflammation induced by IL-1 injected into rabbit joints [59]. Therefore, total GAG is a useful measure to indicate degree of aggrecan breakdown. Another GAG epitope, the serum WF6 epitope, representing a specific pattern of sulfation in chondroitin 6-sulfate was higher in ACL injured compared to healthy controls [60]. In a placebo controlled dog OA model, C2C levels were elevated in the first 4 weeks after induced injury and remained elevated for 16 weeks, indicating increased cartilage turnover leading to onset of OA [54]. C2C can be detected in urine, serum, and synovial fluid and therefore serves as valuable potential marker determining the extent of cartilage loss [61]. Urinary CTXII has the most data supporting its use as a cartilage degradation marker [62]. Recent data suggest urinary CTXII may be most indicative of the turnover in the mineralized cartilage layer [63] and be prognostic for PTOA after ACL injury as decreasing concentrations were associated with decreasing knee pain and improving function [64]. Hyaluronan levels have been shown be predictive of OA progression (summarized in [65] In our pilot trial of ACL injury in humans, synovial fluid IL-1α and serum HA decreased significantly in response to intra-articular IL-1Ra [66]. Other biomarkers are associated with acute joint injury, such as lubricin, stromal cell-derived factor (SDF-1), cartilage intermediate layer protein (CILP), and fibril-associated collagens with interrupted triple helices (FACIT) and fibrillar collagens), but it remains to be seen whether they are predictive of long-term structural damage [12].
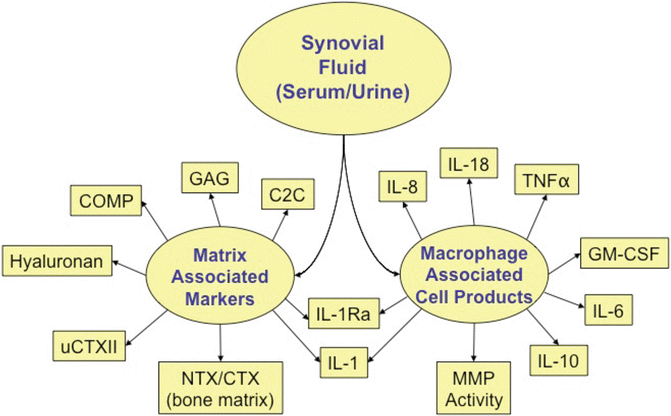
Fig. 25.2
Candidate biomarkers for injury studies. C2C type II collagenase-generated cleavage epitope, COMP cartilage oligomeric matrix protein, GAG glycosaminoglycan, GM-CSF granulocyte-macrophage colony-stimulating factor, IL interleukin, MMP matrix metalloproteinase, NTX/CTX N-terminal telopeptide /C-terminal telopeptide, TNF tumor necrosis factor alpha, uCTXII urinary C-terminal telopeptide of type II collagen
Metabolic Profiling and Metabolites as Biomarkers
Metabolic profiling (metabolomics, metabonomics) is the qualitative and quantitative analysis of small molecules in a system under a given set of conditions (such as healthy or diseased joints). Metabolites are the end-products of cellular regulatory processes, and their levels can be regarded as a global assessment of a cellular state and the ultimate response of biological systems to environmental changes, such as those that might occur in PTOA (intra-articular environment after trauma), taking into account genetic regulation, altered kinetic activity of enzymes, and changes in metabolic reactions [67]. The science of metabolomics was developed decades ago to study inborn errors of metabolism, toxicology, and functional nutrigenomics [67]. More recently, metabolic profiling has been validated as a diagnostic tool and was used to discover citrate and choline as biomarkers for prostate and breast cancer, respectively. In fact, both tests are now covered by health insurance providers [68–70].
The last decade has seen an increase in the use of metabolic profiling as a predictive tool for OA. Lamers et al. [71] used nuclear magnetic resonance (NMR) to study urine from Hartley outbred guinea pigs that spontaneously develop OA. Lactic acid, malic acid, hypoxanthine and alanine were found to contribute heavily to the metabolic profile of OA. In a follow-up patient-based study [72], they demonstrated that the NMR spectra could discriminate between healthy and OA groups. Recently, Zhai et al. [73] employed metabolic profiling on human serum and demonstrated that ratios of valine and leucine to histidine were predictive of OA. While these studies measure metabolites in serum or urine, which are more reflective of systemic arthritis burden, PTOA is joint specific and may more accurately be characterized by a joint-specific profile, such as that found in the synovial fluid. Indeed, metabolic profiling has been performed on synovial fluid from experimentally induced OA in canine knee joints using NMR [74] and demonstrated a hypoxic and acidotic environment with OA that uses fat metabolism as an energy source.
Likewise, Adams et al. [75] performed metabolic profiling on the synovial fluid from patients with end-stage ankle PTOA. Synovial fluid from patients without ankle pathology was used as a healthy control. Metabolic profiling identified 182 metabolites across all synovial fluid samples. Of these, 106 (58 %) were found to be significantly elevated in the PTOA group and one was significantly higher in the control group (threonine). A random forest analysis was performed on the data to determine whether healthy and PTOA samples could be differentiated from one another based on their metabolic profile, and to determine which metabolites were most influential to differentiate between groups. Random forest analysis yielded a predictive accuracy of 90 % when using the metabolic profiles to distinguish between groups. Glutamate, which was >7-fold higher in the PTOA group, ranked number one overall in the random forest analysis and has previously been associated with arthritis [71, 72].
Additionally, significantly elevated levels of proline, trans-4-hydroxyproline, and the dipeptide prolyl-hydroxyproline were found in the PTOA group; these data support increased extra-cellular matrix turnover and collagen breakdown. Evidence of an increased oxidative environment in PTOA was provided by the significantly increased levels observed for oxidized glutathione (GSSH), cysteine disulfide, cystine, cysteine-glutathione disulfide, threonate, and alpha-tocopherol. An increased inflammatory environment in PTOA was evidenced by elevated levels of tryptophan, kynurenine (the inflammatory cytokine-responsive metabolite of tryptophan) and the fibrinogen cleavage peptide DSGEGDFXAEGGGVR, which was significantly higher (>10-fold) in the PTOA group and was the second most important metabolite for differentiating between arthritis and control groups in the random forest analysis. Fibrinogen fragments have been shown to be elevated in the SF of inflamed joints [76], and the citrullinated DSGEGDFXAEGGGVcR peptide has recently been shown to be elevated in patients with rheumatoid arthritis [77]. The kynurenine pathway, which is responsive to immune and inflammatory stimulation, is altered in a variety of human disorders and diseases, including cancer, depression, dementia, and several other neurodegenerative and central nervous system disorders [78–80]. Elevated tryptophan metabolism and kynurenine levels have also been shown in primary synovial cell cultures in response to elevated IFN-γ, suggesting altered or increased tryptophan metabolism in response to inflammatory cytokines associated with arthritis [81]. Additional metabolites including lactate, malate, hypoxanthine, glycerol, isoleucine, hydroxybutyrate, and hydroxyisobutyrate were significantly elevated confirming results from previously reported metabolic studies of synovial fluid [71, 72]. Similar to OA, these results suggest an inflammatory, oxidative, hypoxic and acidotic intra-articular environment for PTOA. These data support the use of metabolites as biomarkers in PTOA.
PTOA Pathogenesis and Timecourse from the Standpoint of Biomarkers
Is PTOA the same as idiopathic OA only more dramatic and with a truncated timecourse? One study of a large cohort of retired National Football League (NFL) players [82] suggests that the two conditions are similar. The overall prevalence of arthritis (mostly OA [87 %]) was higher in retired NFL players (N = 2,538) than in the general United States male population. The retired NFL players likely had PTOA as a consequence of the high incidence of joint injury that occurs with football. Looking at the prevalence of arthritis by age group shows that differences between the retired NFL players and general male population were most apparent at younger ages, but the disparities attenuated with advancing age, and were not significant by age 65+ years. The remarkable similarities in arthritis prevalence in the older age groups of the two populations suggest that PTOA (retired NFL players) and idiopathic, age-related OA (general male population) are comparable conditions, with injury accelerating arthritis onset, but not increasing the occurrence of arthritis across the life course (Fig. 25.3) [82].
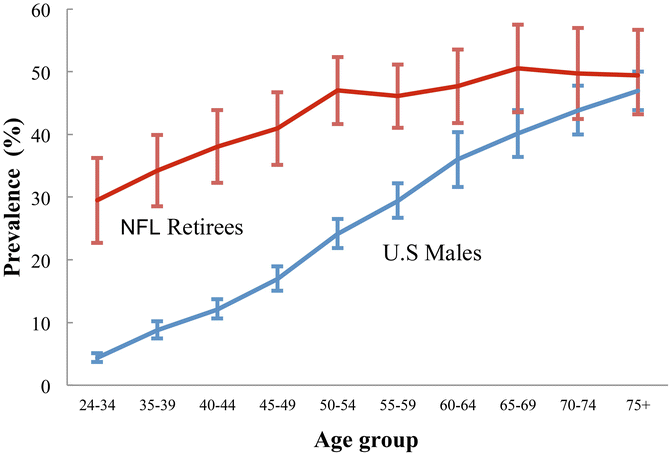
Fig. 25.3
Prevalence of arthritis in retired professional football players and general population of US males. Adapted, by permission, from Y.M. Golightly, S.W. Marshall, L.F. Callahan, and K. Guskiewicz, 2009, “Early-onset arthritis in retired National Football League players,” Journal of Physical Activity and Health 6(5): 648–643 [82]
Conversely, in their 2013 review paper, Little and Hunter [83] stated that PTOA and idiopathic OA may differ by molecular pathophysiology. The authors pointed to the poor translation of preclinical animal studies to Phase II and III human clinical trials, suggesting that the lack of compatibility of the OA animal model (injury-induced in young animals) with human OA (older adults with spontaneous disease) may be a factor in the failure of an effective preclinical intervention in human clinical trials.
Maintenance of cartilage extracellular matrix and suppression of catabolism involves mechanosensing by the transient receptor potential vanilloid 4 (TRPV4), a Ca2+-permeable osmomechano-TRP channel that is highly expressed in articular chondrocytes [84]. Cartilage wounding, as in injury, triggers a wound healing response [85, 86] that is also sensed mechanically although the precise mechanistic details have yet to be fully elucidated. This is made evident by the fact that no OA ensues if the wounded limb is immobilized [87]. The classic wound healing response includes an inflammatory phase, a proliferative phase and a wound remodelling phase with scar tissue formation [88]. Mechanosensing seems key to initiating the inflammatory phase of wound healing.
What do biomarkers tell us about disease pathogenesis in the context of joint injury? For one, PTOA, like OA, appears to involve the innate immune inflammatory response with damage-associated molecular patterns (DAMPs) involved in both [89, 90]. DAMPs are breakdown products of endogenous molecules, such as fibronectin [89] and hyaluronan [90], and molecules released by activated, stressed or dying cells, such as high-mobility group protein 1 (HMGB-1) [90]. DAMPs activate a primitive and powerful innate immune response [91] that accentuates the catabolic response to injury. The surface area of exposed cartilage matrix after fracture is thought to enhance the release of cell debris and inflammatory molecules from the cartilage into the surrounding area, thereby serving as a stimulus for acute and chronic inflammatory processes contributing to the likelihood of PTOA [92]. Therefore, another potentially fruitful area for future development of novel treatments for the prevention of PTOA would be the sequestration of DAMPs [92].
Biomarkers are generally considered indirect measures that may fail to provide reliable evidence about the benefit-to-risk profile of interventions [19]. This would generally be true for biomarkers that are not in the causal pathway of the disease process (indirect biomarkers). However, the mediators of the trajectory to PTOA after joint injury include DAMPs as described above. Many DAMPs can be measured as biomarkers and are directly in the causal pathway of disease (direct biomarkers) (Fig. 25.4). Thus, joint disease is one field in which the development of direct biomarkers appears eminently feasible.
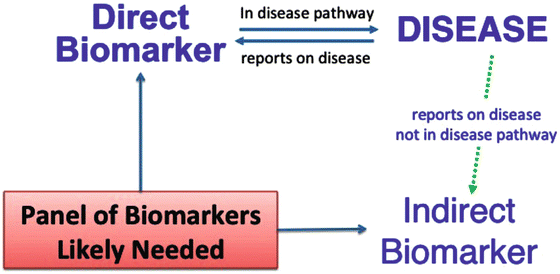
Fig. 25.4
 Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
 Outcomes of ACL Injury: The MOON Consortium
Outcomes of ACL Injury: The MOON Consortium
 Aging and Post-Traumatic Arthritis
Aging and Post-Traumatic Arthritis
 Instability: Dynamic Loading Models
Instability: Dynamic Loading Models




Conceptual diagram of direct and indirect biomarkers of a disease process. Danger signals, in the form of disease associated molecular patterns (DAMPs) are generated in PTOA. These have the potential to report on the disease process as well as activate it through stimulation of pathways of the innate immune system. Although to date, no studies have reported genetic or epigenetic factors associated with susceptibility to PTOA [12], based on results from genetically engineered mice [83] it is likely that a number of genetic modifications provide protection against or worsen PTOA. For instance, the intensity of response of the innate immune system in an individual may in part be controlled by genetic determinants [93]. Indirect biomarkers may report on the disease process but may not be directly involved in disease pathogenesis. Given the complexity and multitude of joint tissues involved in development of PTOA, it is likely that a panel of several, rather than a single biomarker, will be required for adequately predicting susceptibility to PTOA in the pre-injury and post-injury periods
Related posts:
Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
Outcomes of ACL Injury: The MOON Consortium
Aging and Post-Traumatic Arthritis
Instability: Dynamic Loading Models
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
