Basal Ganglia and Neurotransmitter Disorders
Joseph Jankovic
Biochemical or structural pathology in the basal ganglia may be manifested by movement disorders, groups of neurologic diseases, or syndromes that are characterized either by slowness, paucity, and “freezing” of voluntary movement (bradykinesia, akinesia) or by excess abnormal involuntary movement (hyperkinesia, dyskinesia). The basal ganglia seem to be important in the initiation, scaling, and controlling of the amplitude and direction of movement. This complex of deep nuclei is divided anatomically into the corpus striatum, the globus pallidus, and the substantia nigra (Fig. 414.1). The corpus striatum, which includes the caudate nucleus and the putamen, receives input from the cerebral cortex and the thalamus and, in turn, projects to the globus pallidus. The substantia nigra is divided into the dopamine-rich pars compacta, which
is darkly pigmented because of a high content of neuromelanin, and the less dense pars reticularis. The pars reticularis is similar histologically and chemically to the medial segment of the globus pallidus, and both project by way of the thalamus to the premotor and motor cortex. The pars compacta gives rise to the nigral-striatal pathway, which is the main dopaminergic tract. The output of the basal ganglia, which once was thought to be in parallel with the pyramidal system (hence the term extrapyramidal), projects by way of the thalamus to the cerebral cortex and then to the pyramidal system. Integration of the basal ganglia with the cortex facilitates motor control.
is darkly pigmented because of a high content of neuromelanin, and the less dense pars reticularis. The pars reticularis is similar histologically and chemically to the medial segment of the globus pallidus, and both project by way of the thalamus to the premotor and motor cortex. The pars compacta gives rise to the nigral-striatal pathway, which is the main dopaminergic tract. The output of the basal ganglia, which once was thought to be in parallel with the pyramidal system (hence the term extrapyramidal), projects by way of the thalamus to the cerebral cortex and then to the pyramidal system. Integration of the basal ganglia with the cortex facilitates motor control.
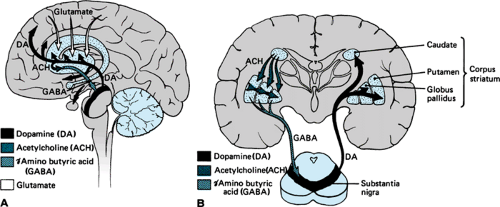 FIGURE 414.1. Brain diagram showing some important neurotransmitter pathways involved in disorders of the basal ganglia. A: Sagittal section. B: Coronal section. |
The diagnosis of a particular movement disorder depends primarily on careful observation of the clinical phenomena. Often, the bradykinetic movement disorders are accompanied by rigidity, postural instability, and loss of automatic associated movements. The hyperkinetic involuntary movements are differentiated phenomenologically according to their characteristic clinical features: rapidity and duration of contractions, rhythmicity, pattern, and suppressibility (Table 414.1). In general, abnormal involuntary movements are exaggerated with stress and disappear during sleep; however, certain forms of myoclonus and tics may persist during all stages of sleep. In a clinic devoted to movement disorders, tics are the hyperkinetic movements observed most commonly in children, followed in lessening frequency by dystonia, stereotypies, choreoathetosis, tremors, and myoclonus.
PARKINSONISM
Parkinson Disease
Parkinson disease (PD) usually is a condition of middle and late life, but the early-onset parkinsonism (before age 40) and juvenile parkinsonism (before age 20) seems to be recognized with increasing frequency. Although the rigid, akinetic form of parkinsonism appears to occur more commonly in the juvenile cases, the typical resting tremor is present in many affected patients. Dystonia (often involving the legs), levodopa-induced dyskinesias, and clinical fluctuations seem to be particularly common occurrences in the juvenile cases. Some patients with familial juvenile parkinsonism belong to group with the “hereditary dystonia-parkinsonism syndrome.” The pathogenesis of PD is further described in Box 414.1. Many other types of parkinsonism, some of which have their onset in childhood (Box 414.1), have been identified.
TABLE 414.1. DIFFERENTIAL DIAGNOSIS OF HYPERKINETIC MOVEMENT DISORDERS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Wilson Disease
Wilson disease (WD) is one of the most important causes of juvenile parkinsonism and other movement disorders. Failure to
diagnose this condition may have tragic consequences, including death, as a result of irreversible liver cirrhosis and profound neurologic deficit. The gene for this autosomal recessive hepatolenticular degenerative disease has been linked to the esterase D locus on the long arm of chromosome 13. Numerous mutations have been identified in the WD gene, which encodes copper-transporting P-type adenosine triphosphatase (ATPase) and has been termed ATP7B. The prevalence of the gene in the overall population is approximately 1%, although symptomatic WD is a relatively rare event (estimated prevalence, 30 per 1 million). The hepatic and neurologic dysfunction associated with WD is caused by a defect in copper metabolism, resulting from a reduction in the rate at which copper is incorporated into ceruloplasmin and a decrease in the rate at which it is excreted from the liver. Usually, low ceruloplasmin levels are found in patients with WD, but they do not seem to be the primary defect because some patients with the disease and some heterozygotes have normal levels. Furthermore, the ceruloplasmin gene has been mapped to chromosome 3, not to chromosome 13. The excess copper not only accumulates in the liver and brain but also can cause renal tubule damage, osteoporosis, and arthropathy. Kayser-Fleischer rings, the best-recognized ophthalmologic sign of WD, are caused by the deposition of copper in the cornea.
diagnose this condition may have tragic consequences, including death, as a result of irreversible liver cirrhosis and profound neurologic deficit. The gene for this autosomal recessive hepatolenticular degenerative disease has been linked to the esterase D locus on the long arm of chromosome 13. Numerous mutations have been identified in the WD gene, which encodes copper-transporting P-type adenosine triphosphatase (ATPase) and has been termed ATP7B. The prevalence of the gene in the overall population is approximately 1%, although symptomatic WD is a relatively rare event (estimated prevalence, 30 per 1 million). The hepatic and neurologic dysfunction associated with WD is caused by a defect in copper metabolism, resulting from a reduction in the rate at which copper is incorporated into ceruloplasmin and a decrease in the rate at which it is excreted from the liver. Usually, low ceruloplasmin levels are found in patients with WD, but they do not seem to be the primary defect because some patients with the disease and some heterozygotes have normal levels. Furthermore, the ceruloplasmin gene has been mapped to chromosome 3, not to chromosome 13. The excess copper not only accumulates in the liver and brain but also can cause renal tubule damage, osteoporosis, and arthropathy. Kayser-Fleischer rings, the best-recognized ophthalmologic sign of WD, are caused by the deposition of copper in the cornea.
Usually, the onset of WD occurs between adolescence and age 40, and its presentation seems to be age-dependent, with hepatic failure being more common in children and neurologic or psychiatric symptoms occurring frequently in adults. In a nonselected population of 31 patients with WD, the mean age at onset was 21±5 years; 61% had neurologic symptoms, 13% had liver symptoms, and 10% had a combination of neurologic and liver problems. Approximately one-third of patients
had psychiatric symptoms, including depression, emotional lability, personality change, and slow mentation, at onset of disease. Another one-third had neurologic symptoms, particularly parkinsonism, pseudobulbar palsy, tremor, and dystonia; 14% had symptoms of liver disease; and 17% had their condition diagnosed by family screening. The most common neurologic findings at first evaluation were dysarthria (97%), dystonia (65%), dysdiadochokinesia (58%), rigidity (52%), gait and postural abnormalities (42%), tremor (32%), abnormal eye movements (32%), hyperreflexia (29%), drooling (23%), and bradykinesia (19%).
had psychiatric symptoms, including depression, emotional lability, personality change, and slow mentation, at onset of disease. Another one-third had neurologic symptoms, particularly parkinsonism, pseudobulbar palsy, tremor, and dystonia; 14% had symptoms of liver disease; and 17% had their condition diagnosed by family screening. The most common neurologic findings at first evaluation were dysarthria (97%), dystonia (65%), dysdiadochokinesia (58%), rigidity (52%), gait and postural abnormalities (42%), tremor (32%), abnormal eye movements (32%), hyperreflexia (29%), drooling (23%), and bradykinesia (19%).
BOX 414.1 Classification of Parkinsonism
Primary Parkinson Disease
Idiopathic, dominated by:
Tremor
Postural instability, gait difficulty
Akinesia (freezing)
Dementia
Depression
Sensory disturbance
Autonomic dysfunction
Inherited, associated with essential tremor, dystonia, or peripheral neuropathy
Young-onset, associated with dystonia or essential tremor
Secondary Parkinsonism
Drugs (dopamine-blocking and -depleting drugs, alpha-methyldopa, lithium, diazoxide, flunarizine, cinnarizine)
Toxins (manganese, mercury, carbon monoxide, cyanide, carbon disulfide, methanol, ethanol, MPTP)
Metabolic (parathyroid, acquired hepatocerebral degeneration, GM1 gangliosidosis, Gaucher disease)
Encephalitis and postencephalitic syndrome
Slow virus (Creutzfeldt-Jakob disease)
Vascular (multiinfarct, Binswanger disease)
Brain tumor
Trauma and pugilistic encephalopathy
Hydrocephalus (normal and high-pressure)
Syringomesencephalia
Multiple System Degenerations (Parkinsonism Plus)
Sporadic
Progressive supranuclear palsy (ophthalmoparesis)
Shy-Drager syndrome (dysautonomia)
Olivopontocerebellar atrophy (ataxia)
Parkinsonism-dementia-amyotrophic lateral sclerosis complex
Striatonigral degeneration
Corticodentonigra degeneration with neuronal achromasia
Alzheimer disease
Inherited
Huntington disease
Wilson disease
Hallervorden-Spatz disease
Familial parkinsonism-dementia syndrome
Familial basal ganglia calcification
Neuroacanthocytosis
Spinocerebellar-nigral degeneration and Joseph disease
Glutamate dehydrogenase deficiency
Footnote
MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
Modified with permission from
Jankovic J. Parkinson’s disease and related disorders of movements. In: Calne DB, ed. Handbook of experimental pharmacology. Berlin: Springer-Verlag, 1989: 227.
Because of its variable clinical expression, establishing the diagnosis of WD often is delayed. Almost all patients with neurologic disease have Kayser-Fleischer rings, but this yellow-brown deposit at the limbus of the cornea may be noted also in patients with primary biliary cirrhosis and active hepatitis with cirrhosis. Occasionally, a “sunflower cataract” may be seen as a result of copper deposition in the lens.
Usually, the diagnosis of WD is confirmed by the demonstration of low serum copper and ceruloplasmin levels, increased urine copper concentrations after a dose of penicillamine, and a rise in the level of copper in the liver (Table 414.2). The ratio of radioactivity in the plasma at 24 hours to that at 2 hours after the administration of an oral or intravenous dose of copper64 is less than 0.5 in patients with WD (the equivalent value being greater than 0.8 in normal individuals). Other laboratory abnormalities often detected in patients with WD include aminoaciduria, hypercalciuria, glycosuria, leukopenia, hemolytic anemia, thrombocytopenia, and renal tubal deficit. In approximately one-half of affected patients, computed tomographic (CT) scanning of the brain reveals characteristic hypodense areas in the region of the basal ganglia, and often magnetic resonance imaging (MRI) reveals on T2-weighted images hypointensity that extends from the globus pallidum into the putamen. Extensive degeneration of the corpus striatum, particularly the putamen, is seen at autopsy. Also, the cerebellum, brainstem, and subcortical white matter may be involved.
The goals of treating patients with WD are to reduce copper intake and to create a negative copper balance by increasing copper output in the urine. D-Penicillamine at dosages of 0.5 to 2.0 g/day has been used successfully to chelate copper. However, penicillamine often produces considerable toxicity, including fever, urticaria, leukopenia, nephritis, thrombocytopenia, systemic lupus erythematosus, hemolytic anemia, Goodpasture syndrome, pyridoxine deficiency, and a syndrome resembling myasthenia gravis. Trientine hydrochloride (Syprine) in dosages of 400 to 800 mg three times daily before meals has been approved for the treatment of penicillamine-sensitive patients with WD. In addition to the chelating agents, zinc sulfate at a dosage of 300 to 1,200 mg/day between meals may reduce the absorption of copper. When early diagnosis is made and dietary and chelating therapies are instituted, the progression of liver and neurologic dysfunction can be halted and even reversed. At least 2,000 μg of copper should be excreted during the first 24 hours of penicillamine therapy. Approximately one-third of patients with WD experience deterioration despite receiving penicillamine therapy, but most improve, sometimes after a latent period lasting several weeks or months. In patients with parkinsonian symptoms, levodopa and anticholinergic therapy may provide symptomatic relief.
Besides the treatment of patients with symptomatic WD, of paramount importance is screening all their relatives and instituting therapy immediately if the disease is diagnosed in any of them. Patients who fail to improve with chelating or other pharmacologic therapy may experience marked relief of neurologic symptoms several months after undergoing liver transplantation.
Huntington Disease
The usual onset of Huntington disease (HD) occurs in the fourth and fifth decades of life, but it is seen in approximately
10% of affected patients during childhood or adolescence. Both juvenile and adult-onset HD are autosomal dominant traits, with a defective gene mapped to a terminal band of the short arm of chromosome 4. The gene mutation was found to consist of an unstable enlargement of the CAG repeat sequence at 4p16.3. Most patients with juvenile HD have the akinetic-rigid syndrome termed the Westphal variant. Other features of juvenile HD include dementia, seizures, and ataxia. In addition, patients with juvenile HD are more likely to have inherited the abnormal gene from their father than from their mother, and they tend to segregate within families. Although the medium-sized spiny neurons (type I cells) usually are affected first in the adult form of HD, the large spiny cells have been suggested to degenerate first in the juvenile form. In contrast to the caudate nucleus, which typically is involved in the adult form of HD, the putamen seems to be most damaged in the juvenile form of the disease.
10% of affected patients during childhood or adolescence. Both juvenile and adult-onset HD are autosomal dominant traits, with a defective gene mapped to a terminal band of the short arm of chromosome 4. The gene mutation was found to consist of an unstable enlargement of the CAG repeat sequence at 4p16.3. Most patients with juvenile HD have the akinetic-rigid syndrome termed the Westphal variant. Other features of juvenile HD include dementia, seizures, and ataxia. In addition, patients with juvenile HD are more likely to have inherited the abnormal gene from their father than from their mother, and they tend to segregate within families. Although the medium-sized spiny neurons (type I cells) usually are affected first in the adult form of HD, the large spiny cells have been suggested to degenerate first in the juvenile form. In contrast to the caudate nucleus, which typically is involved in the adult form of HD, the putamen seems to be most damaged in the juvenile form of the disease.
TABLE 414.2. LABORATORY TESTS IN WILSON DISEASE | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The diagnosis of HD can be established with 100% accuracy with the demonstration of the presence of more than 40 CAG repeats in one of the alleles in the HD gene. The psychological and social impact of such testing, however, must be considered carefully before presymptomatic individuals are tested.
The most remarkable biochemical change observed in the brains of adults with HD is a reduction in the activity of glutamic acid decarboxylase, particularly in the corpus striatum, substantia nigra, and other basal ganglia. In contrast, thyrotropin-releasing hormone, neurotensin, somatostatin, and neuropeptide Y are increased in the corpus striatum. The depletion of gamma-aminobutyric acid in the corpus striatum may result in disinhibition of the nigral-striatal pathway. Coupled with the accumulation of somatostatin, the net result may be the release of striatal dopamine, which results in chorea. Dopamine-blocking drugs, such as haloperidol, and dopamine-depleting agents, including tetrabenazine, often are useful in controlling chorea. In patients with childhood HD, which usually is manifested by parkinsonian features, levodopa may provide symptomatic relief.
Hallervorden-Spatz Disease
In contrast to WD, which is characterized by the abnormal deposition of copper, the hallmark of neurodegeneration with brain iron accumulation (NBIA), previously referred to as Hallervorden-Spatz disease, is the deposition of iron, particularly in the globus pallidus and substantia nigra. Usually, NBIA starts in patients between the ages of 4 and 12 years, but occasionally it presents as parkinsonian dementia in adult life. Children with NBIA have posture and gait abnormalities, bradykinesia, rigidity, and other parkinsonian features, including tremor. In addition, other hyperkinetic movement disorders, particularly dystonia and choreoathetosis, may be seen. Further symptoms include progressive dysarthria, dementia, ataxia, spasticity, seizure disorder, optic atrophy, and retinitis pigmentosa.
Most cases are inherited in an autosomal recessive pattern, but many occur sporadically, and some phenotypically similar cases seem to be inherited by autosomal dominant transmission. Mutations in the gene PANK2, localized to 20p12.3–p13, have been identified in a subset of patients with NBIA, referred to as pantothenate kinase-associated neurodegeneration (PKAN). Testing for the most frequent mutations has been used to confirm the diagnosis of PKAN. The disease also may be suspected when a MRI scan shows a central focus of increased T2-signal intensity surrounded by a zone of decreased signal in the region of the globus pallidus (eye-of-the-tiger sign) (Fig. 414.2).
Furthermore, increased iron uptake in the basal ganglia may be demonstrated by scintillation counting after the infusion of radioactive iron (59Fe). Increased iron uptake is confirmed also by postmortem examination, which reveals pigmentary degeneration of the basal ganglia, particularly the internal segment of the globus pallidus and the zona reticularis of the substantia nigra. These pigmentary changes are caused by a threefold to fourfold accumulation of iron in these areas. Another distinctive pathologic feature of NBIA is marked neuroaxonal degeneration, with the formation of spheroids. These glycoprotein-containing axonal swellings have been attributed to abnormal peroxidation. The abnormal accumulation of cysteine was demonstrated in one of our patients with NBIA. This increased cysteine may chelate ferrous iron, which in turn promotes the generation of free radicals, causing the characteristic neuropathologic changes of the disease. Iron chelation with desferrioxamine and antioxidant therapy has been tried in patients with NBIA, but no benefit has been demonstrated.
Levodopa and anticholinergic drugs may provide modest symptomatic relief of parkinsonian symptoms. In addition, botulinum toxin injections usually are effective in the treatment of focal dystonia (see the section, Dystonia).
HYPERKINETIC MOVEMENT DISORDERS
Tremor
Essential tremor (ET) is the most common cause of an oscillatory involuntary movement during childhood (Box 414.2). ET may start at any age, including infancy. One form of infantile ET is the hereditary chin tremor, which consists of rhythmic,
three-per-second contractions of the chin that often are associated with deafness and are inherited in an autosomal dominant pattern. Another form of ET that begins during infancy or early childhood is so-called shuddering attacks. Affected children may have more than 100 attacks a day, but symptom-free intervals may last as long as 2 weeks. The attacks are characterized by bursts of rapid trembling of the entire body, occasionally associated with head turning, involuntary sniffing, and throat clearing. During the attacks, affected children usually sink to the floor; the attacks may persist during sleep.
three-per-second contractions of the chin that often are associated with deafness and are inherited in an autosomal dominant pattern. Another form of ET that begins during infancy or early childhood is so-called shuddering attacks. Affected children may have more than 100 attacks a day, but symptom-free intervals may last as long as 2 weeks. The attacks are characterized by bursts of rapid trembling of the entire body, occasionally associated with head turning, involuntary sniffing, and throat clearing. During the attacks, affected children usually sink to the floor; the attacks may persist during sleep.
In addition to these forms of ET, the characteristic action-postural tremor also may be seen in children. Often, the slower tremor (approximately 6.5 Hz) involves the head and neck, whereas the more rapid tremor (8 to 12 Hz) tends to involve the hands. Many other variants of ET have been recognized, however. Although ET usually is “benign,” it occasionally can progress to a very disabling movement disorder, interfering with writing, feeding, speaking, and other activities of daily living.
Usually, ET is inherited in an autosomal dominant manner, and a locus has been found on chromosome 2p22–p25. Although abnormalities of neurotransmitters in the basal ganglia are suspected to underlie ET, no pathologic changes have been documented in the few brains that have been examined at autopsy. Besides the beta blockers, primidone, gabapentin, topiramate, lorazepam, alprazolam, clonazepam, amantadine, clonidine, and ethanol may improve ET.
Other oscillatory involuntary movements occasionally seen in infants and children are “head nodding,” which often is associated with congenital nystagmus, including spasmus nutans, and the “bobble-headed doll’s syndrome,” which is seen with diencephalic lesions, including third-ventricle cysts or tumors, craniopharyngioma, hydrocephalus, and hypothalamic lesions.
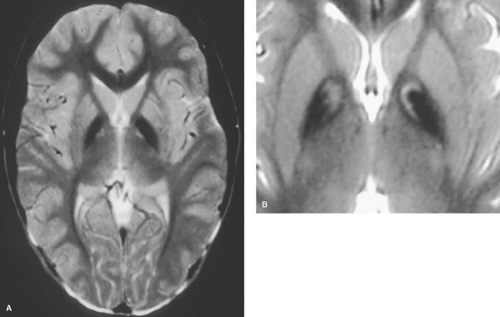 FIGURE 414.2. MRI of a patient with NBIA showing hypointensity with relative hyperintensity in the anteromedial globus pallidus on T2-weighted MRI, the so-called “eye-of-the-tiger” sign.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
