Fig. 14.1
Systemic aging changes that could contribute to the development of osteoarthritis, including post-traumatic OA. Reprinted with permission from HSS Journal 2012; 8:19
Relationship Between Age and the Development of PTOA
OA is a slowly progressive chronic disease that develops over years. Therefore, the classic OA pathological and radiological findings (cartilage destruction, subchondral bone thickening, and osteophytes) are not seen immediately after a joint injury but rather take time to develop. The influence of time on the development of OA allows for aging to contribute to the development of PTOA. In a longitudinal study of former medical students who experienced a knee injury at a mean age of 22 years, the cumulative incidence of clinically diagnosed knee OA by 65 years of age was 13.9 % compared to 6.0 % who did not report an injury [7]. In that study, which followed a young cohort, the average time to symptomatic OA after the knee injury was 22 ± 13 years with the majority of the subjects not having OA diagnosed until after 45–50 years of age.
The time frame between injury and the diagnosis of OA appears to vary from person to person due to a number of factors that include the age at which the individual experienced the joint injury as well as the type of injury. Risk factors for noninjury-related OA, such as lower extremity malalignment, obesity, female gender, joint overuse, and certain genetic polymorphisms, are also likely to accelerate the onset of PTOA and account for the variability in this patient population. In a retrospective cross-sectional study of adults with injuries to the anterior cruciate ligament (ACL) or the meniscus, Roos et al. [8] reported that radiographic OA appeared on average at 10 years after the injury. However, the time frame varied by patient age such that those who had an isolated meniscus injury between the ages of 17 and 30 took about 15 years to develop radiographic OA, while patients over the age of 30 when the injury occurred took only about 5 years.
Attempting to study the relationship between aging and PTOA in cross-sectional studies is complicated by the length of time it takes for OA to develop after an injury. Because of this, older adults may not recall an injury that occurred earlier in life or when it occurred. MRI studies of older adults with symptomatic OA commonly find evidence of ACL tears and/or meniscal tears in people who do not recall having a joint injury [9–11]. These tears may have resulted from relatively minor injuries or from aging changes in joint tissues that predispose to nontraumatic or “degenerative” tears. In women over the age of 40 with knee OA, 73 % were found to have a meniscus tear by MRI [11], and in a group of male and female subjects with an average age of 67 years, 22.8 % had a complete ACL rupture [10]. Even in a Korean cohort with an average age of 71 years randomly chosen regardless of knee OA, meniscal and ligament damage was present in 49.75 and 8 % of men and 71.2 and 26.9 % of women, respectively [9].
Studies have begun to investigate mechanisms for the relationship between meniscal injury and the development of OA. The meniscus is a structure vital to the normally functioning knee joint that contributes to shock absorption, joint stability, articular congruity, and proprioception [12]. Loss of meniscus function has detrimental effects on the knee joint [13–16]. Total meniscectomy reduces knee joint contact area by 50 % and greatly increases the load on the knee with subsequent damage and degeneration of articular cartilage and development of OA [17]. Even partial meniscectomy significantly increases contact pressures and leads to increased joint wear [13].
Meniscus tears in non-osteoarthritis subjects are felt to be an early event in the OA disease process and may be a risk factor for subsequent articular cartilage degeneration [18]. Meniscus tears or degeneration noted by MRI was found to be significant predictors for the development of OA 30 months later in middle-aged and elderly individuals (between 50 and 79 years of age) with an odds ratio of 5.7. Meniscus extrusion on MRI, which is consistent with diminished hoop mechanical function of the meniscus, has been shown to be associated with loss of cartilage volume over 2 years mediated by subchondral bone changes (bone expansion and cartilage loss) [18]. Furthermore, body mass index, tibial bone area, joint alignment, past knee injury, and presence of osteophytes may be causally related to the development of meniscus extrusion [18, 19].
In addition to altered joint mechanics, the injured meniscus has also been suggested to play a biologic role in the development of knee OA [20–23]. A study of gene expression in human meniscus tissue, removed at the time of partial meniscectomy after a meniscal injury with or without an ACL injury, compared individuals younger or older than 40 years [20]. The younger individuals appeared to have a greater response to the injury with higher expression levels of interleukin (IL)-1β and several matrix-degrading enzymes including a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-5; matrix metalloproteinase (MMP)-1, MMP-9, and MMP-13; as well as NFκB2 [20]. The same group also reported results of a microarray study, again using samples from partial meniscectomy, and correlated the results with age and degree of cartilage damage [23]. Using a cutoff difference of ≥1.5-fold, 866 genes were found to be differentially regulated in individuals equal to or younger vs. older than 40 years.
Studies in a mouse model of PTOA have shown an influence of age on the severity of OA. The destabilized medial meniscus (DMM) model is a commonly used model for OA in mice. Histological features of OA at 8 weeks after surgery were found to be twice as severe when DMM surgery was performed on male C57BL/6 mice at 12 months of age compared to 12 weeks of age [24]. In that study, age-associated differences in gene expression were evaluated using microarrays which revealed an age-related decline in expression of matrix genes that included aggrecan and types II and IX collagen in the sham control joints accompanied by an increase in immune and defense response genes. In the DMM joints, more genes (251 vs. 52) were significantly upregulated in the older mice compared to the younger. Genes expressed at higher levels in the older adult mice included ADAMTS-5, asporin, and the chemokine receptors CXCR2 and CXCR7 as well as type III collagen, among others. Younger mice had a greater increase in genes related to the immune response. These results, albeit in a mouse model, demonstrate that the response to joint injury differs significantly between younger and older individuals. The aging changes in joint tissues that might be responsible for these differences will be discussed next.
Aging Changes in Joint Tissues and the Development of PTOA
Cartilage
Among the different joint tissues, cartilage has been most closely examined for changes that occur during the aging process. These changes include alterations in cartilage cells and extracellular matrix. The overall pattern can be described as “chondropenia” and features a reduction in cartilage ECM and cartilage cells [25–35].
Conceptually, loss of chondrocytes could lead to decreased production and maintenance of ECM. Conversely, loss of ECM could expose chondrocytes to abnormal biomechanical stress, leading to changes in their biosynthetic function and cell death. Consequently, age-related changes in the cell and the ECM are closely linked and may potentiate each other. As these changes progress with advancing age, an acute superimposed joint injury can be expected to lead to symptomatic and structurally overt OA more rapidly in older individuals.
Changes in Cartilage ECM
Age-related changes in cartilage ECM include increased cross-linking between collagen fibers and glycosaminoglycans [36, 37] and changes in size and sulfation of aggrecan [38–41]. With aging, there is also an accumulation of advanced glycation end products (AGEs) that are formed by nonenzymatic glycation of cartilage proteins, which are prone to this modification due to their slow turnover [42]. Consequences of AGE accumulation are increased stiffness and increased susceptibility to fatigue failure [43, 44]. AGEs also stimulate chondrocytes via binding to the receptor for advanced glycation end products (RAGEs) to produce inflammatory cytokines and MMPs [45–47]. The deposition of calcium pyrophosphate and basic calcium phosphate crystals is very common in aging cartilage [48], and these crystals promote inflammation and degradation of cartilage ECM [49]. Collectively, these ECM changes lead to deterioration in cartilage biomechanical properties [40, 50, 51]. The confined compression modulus of full-thickness cartilage from the patella decreases by ~33 % with age and degeneration grade [52]. Cartilage tensile properties change dramatically with aging in the human knee joint. With increasing age, tensile modulus and strength of human articular cartilage from the distal femur are reduced by about fourfold [53].
Lubrication at the cartilage surface zone is an essential mechanism that protects the tissue from damage caused by friction, heat, and wear. The cells in the superficial zone produce lubricin also called superficial zone protein (SZP) [54], a highly glycosylated protein that functions as a boundary lubricant [55]. With aging, there is a reduction in cellularity in the superficial zone [56] and consequently in the cell sources of lubricin. Low SZP may be one mechanism that accounts for the earliest occurrence of cartilage structural lesions at the cartilage surface.
Changes in Cartilage Cells
Age-related changes in cartilage cells (chondrocytes) include increased cell death and abnormal biosynthetic function. Analyses of human and animal tissues have shown increased levels of apoptosis in aged cartilage compared with mature cartilage, suggesting that apoptosis, or programmed cell death, may be responsible for chondrocyte hypocellularity during the aging process [57–60]. A controlled study in rabbits showed that age predisposes articular cartilage to a reduction in viable cell density and increased expression levels of proapoptotic genes, and this was observed in cartilage that showed no prior signs of OA [58]. Aged rabbits with macroscopically normal cartilage have higher rates of expression of apoptotic genes, including Fas, FasL, and caspase 8, compared with mature rabbits with a similar grade of cartilage [61]. Additionally, MMP-1 was increased in normal aged cartilage compared with normal mature cartilage [61]. These changes may contribute to the observations in both humans and animals, that aging results in a loss of proteoglycans within the ECM and thinning of the articular cartilage [33, 34, 62, 63]. Overall, these results suggest that although aged cartilage may appear grossly normal, microscopic and molecular changes have occurred, including increased expression of apoptotic and matrix-degrading genes with loss of viable chondrocytes and ECM, which may predispose older individuals to more rapid development of OA following joint injury.
In chondrocytes, aging also leads to a decreased sensitivity to critical anabolic factors, such as insulin-like growth factor (IGF-1). A key function of IGF-1 is as a potent inhibitor of apoptosis in many cell types, including chondrocytes [64] as well as a stimulus of ECM synthesis and antioxidants. In OA, there is an increased expression of TRB3, an Akt inhibitor that inhibits the capacity of IGF-1 to promote proteoglycan synthesis and viability [65]. Aging of chondrocytes is also associated with increased production of reactive oxygen species (ROS) [66, 67], decreased ECM synthesis [68], and increased secretion of matrix-degrading enzymes [69]. Elevated intracellular ROS have been linked to apoptosis and catabolic activity in chondrocytes [70–73]. Because both joint injury and aging can contribute to increased production of ROS, oxidative stress may be an important mechanism by which aging contributes to the development of PTOA (Fig. 14.2).
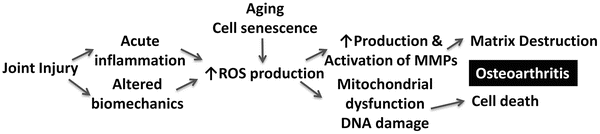
Fig. 14.2
Hypothetical mechanism for an interaction between joint injury and aging leading to osteoarthritis due to excessive production of reactive oxygen species (ROS)
Mechanisms of Cartilage Cell Aging
Several mechanisms that contribute to aging-associated cartilage cell dysfunction and death have been identified. There is evidence for senescence in at least certain subsets of cartilage cells [74], and this is associated with an abnormal biosynthetic activity termed senescence-associated secretory phenotype (SASP) [75]. This includes IL-1, IL-6, IL-7, IL-8, GROα, MCP-2, and MMP-3, which are also produced by OA chondrocytes. In human OA cartilage, RNA levels of MMP-1, MMP-8, and MMP-13 and tissue inhibitor of metallproteinase-1 were altered in cells isolated from lesion sites, where telomere shortening and the senescence marker senescence-associated beta galactosidase were observed, but also in sites distal to the lesions where a lower number of cells exhibited the senescence-like changes [76].
Mechanisms that protect against aging-associated cell damage include autophagy and the unfolded protein response (UPR). Autophagy is a process that is responsible for the removal of damaged cellular organelles such as damaged mitochondria and macromolecules [77]. Conceptually, autophagy in normal adult articular cartilage is an important mechanism for cellular homeostasis. Cells in the normal cartilage superficial zone show stronger expression of autophagy proteins, such as beclin-1, Atg5, and LC3 [78], as compared to cells in the mid and deep zone, and their expression also differs between cartilage regions that are exposed to high versus low levels of mechanical load. Under normal conditions, there is a clearly detectable level of autophagy activation in healthy mouse joints. The highest levels were observed in the chondrocytes present in the superficial and upper middle zone of the articular cartilage in knee joints [79]. In contrast, only few cells in the deep zone contained detectable levels of autophagosomes. As with other tissues, starvation increased the number of autophagosomes in chondrocytes [79].
During the aging process in mouse and human knee joints, there is a decrease in Ulk1, LC3, and beclin-1 protein expression in articular cartilage. The reduction of these key regulators of autophagy is accompanied by increased apoptosis [78]. In a rapidly progressing experimental OA model in mice induced by surgical knee destabilization (DMM surgery plus medial collateral ligament transection), which can be considered as a model of PTOA, there is also a time-dependent reduction in these autophagy proteins [78]. Since this was observed in relatively young mice, it is apparently not a consequence of aging-related mechanisms but related to excessive mechanical load. However, for both surgical OA and mechanically injured cartilage, the increased cell death suggests that dysfunctional autophagy may contribute to cell death.
When full-thickness cartilage explants are exposed to single high impact mechanical compression, there are immediate matrix changes and a low level of cell death. It was observed that there was a short and transient increase in the levels of LC3-II, followed by a marked reduction in the levels of several autophagy proteins, including Ulk1, LC3, and beclin-1 [80]. Thus, during the development of OA, autophagy may increase as an adaptive response to protect cells from various stresses, and failure of the adaptation may lead to further progression of degeneration. The reduction in autophagy protein levels and autophagy activation supports the hypothesis that the basal autophagic activity decreases with the age, thus contributing to the accumulation of damaged organelles and macromolecules and susceptibility to aging-related diseases [81]. A series of prior studies demonstrated mitochondrial dysfunction in various OA models and in human OA [82]. In addition, mitochondrial DNA mutations are increased in OA chondrocytes [83]. Damaged mitochondria, producing high levels of ROS, have an important role in pro-inflammatory signaling, as they initiate formation of inflammasomes and activation of other inflammatory pathways [84].
Autophagy is also involved in regulating differentiation and gene expression. In human knee chondrocytes, IL-1 or nitric oxide increased expression of LC3 and beclin-1 and activated autophagy [85]. Further, autophagy activation prevented IL-1-mediated suppression of cartilage matrix degradation and reduced levels of MMP-13, ADAMTS5, and ROS. Given that one of the cytoprotective functions of autophagy is the removal of damaged mitochondria [83], the IL-1-induced OA-like gene expression changes might possibly occur through reduction of the intracellular ROS level via elimination of damaged mitochondria.
The UPR is normally a repair response that sends simultaneous survival, death, and inflammatory signals and culminates in coordinate transcriptional and translational reprogramming to modulate protein folding, inflammation, and cell fate [86]. Both autophagy and UPR are compromised in aging cartilage, and this contributes to increase oxygen radical production, DNA damage, and cell death. In OA cartilage, increased XBP1 activation and expression of GRP78 and C/EBP homologous protein (CHOP) are evidence of heightened endoplasmic reticulum (ER) stress in situ [87, 88]. Cultured OA chondrocytes demonstrate evidence of PERK module activation [89, 90]. This includes expression of TRB3, an Akt inhibitor that inhibits the capacity of IGF-I to promote PG synthesis and viability. Excess UPR activation can promote chondrocyte hypertrophy or apoptosis, thereby potentially accelerating OA [89, 90]. Excess CHOP expression is one feature of UPR and ER stress. UPR activation was increased in human knee OA cartilage in situ and in biomechanically injured cultured chondrocytes in vitro [91]. In normal human chondrocytes, CHOP “gain of function” sensitized chondrocytes to IL-1β-induced nitric oxide and MMP-3 release without inducing these responses by itself. Excess CHOP expression, by itself, induced superoxide production and apoptosis.
Aging-Related Differences in Cartilage Response to Injury in Experimental Models
A limited number of in vitro and animal model studies have directly addressed age-related changes in cartilage tissue and the cell response to injury. Injurious loading elevates secretion of matrix-degrading enzymes, inflammatory cytokines, and oxygen radicals and causes chondrocyte death [92]. In the ACL transection model, aged rabbits showed faster and more severe cartilage degradation than mature rabbits. Aged rabbits with macroscopically normal cartilage had higher rates of expression of apoptotic genes, including Fas, FasL, caspase 8, and p53 compared with mature rabbits, and this correlated with decreased chondrocyte numbers [58, 61]. As detailed above, experimental OA is also more severe in older as compared to young mice [24]. Gene expression analysis of entire knee joints showed that there were significant age-related differences in gene expression in the sham-operated knees, which included reduced ECM genes, Sox9, and TGFß2 and increased inflammatory cytokine genes. Following knee destabilization, a significantly larger number of genes were upregulated in the older mice, potentially indicative of a more active disease process [24].
These studies in animal models show that experimental OA that is induced by chronic mechanical injury is more severe in older animals. Preexisting changes in gene expression and a different response of the older tissues to injury appear to determine increased tissue damage.
Meniscus
Age-related changes in the meniscus that contribute to OA are likely due to the complex interplay of changes in cellularity, vascularity, and the extracellular matrix in addition to changes in gene expression and senescence (Fig. 14.3). The meniscus is a fibrocartilaginous tissue with a heterogeneous cell composition. The fibrochondrocyte, the cell type populating the meniscus, has two main phenotypic forms: elongated fibroblast-type cells found in the outer meniscus and more rounded chondrocyte-like cells found in the inner meniscus [93–95]. Research involving meniscus explant cultures has identified regional differences between the inner and outer zone cell populations in response to growth factors such as platelet-derived growth factor which correlate with the greater healing potential in the outer zone which is the vascular region of the meniscus [96, 97]. Interestingly, regional differences in healing between inner and outer zone cell populations are maintained in organ culture models, without the active presence of a peripheral vascular supply, suggesting some component of intrinsic healing present in outer zone meniscus cells that is separate from the variable of blood supply [96].
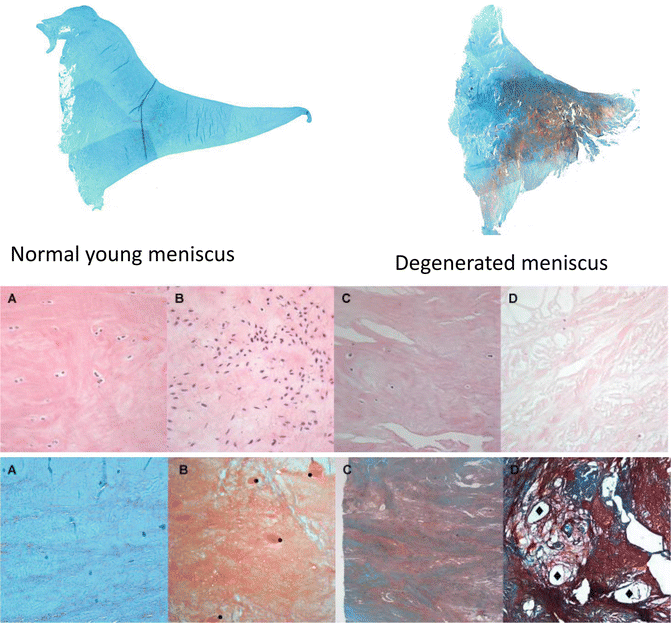
Fig. 14.3
Aging-associated changes in meniscus. Menisci were collected from human knees at autopsy. Representative samples were selected to illustrate different stages and severities of aging-associated changes. Top panel shows cross sections of human medial meniscus stained with safranin O. Middle panels show hematoxylin- and eosin-stained sections of human meniscus at varying degrees of degeneration to illustrate changes in cell density and organization: (a) normal, (b) diffuse hypercellularity, (c) diffuse hypo-/acellular regions, (d) hypocellularity, empty lacunae, and pycnotic cells. Bottom panels show safranin O-stained sections of human meniscus at varying degrees of degeneration to illustrate changes ECM organization. (a) Normal meniscus, collagen fibers organized, homogenous eosinophilic staining of ECM; (b) collagen fibers organized, diffuse foci of hyaline or mucinous degeneration; (c) collagen fibers unorganized, confluent foci or bands of hyaline or mucinous degeneration, fraying; (d) collagen fibers unorganized, fibrocartilaginous separation (edema, cyst formation), severe fraying and tears
Changes in Meniscus Cells
Age has been further shown in meniscus fibrochondrocyte culture studies to be an independent factor contributing to tissue repair. In an inherently avascular system, cultured fetal and juvenile fibrochondrocytes have been shown to have superior repair capacity to adult fibrochondrocytes [98]. Additional cell culture studies have shown that although total collagen production between inner and outer zone populations remains similar, inner zone populations produce significantly higher levels of proteoglycan and exhibit a more chondrogenic phenotype than outer zone cells [99]. Diminished cell density with acellular regions has been noted with age, most dominant in the inner regions of the meniscus [100]. Diminished cellularity is present in tears of meniscus tissue in patients >40 years making that tissue potentially more vulnerable to degeneration and retear following repair compared with younger tissue [101].
Changes in Meniscus Vascularity
Changes in vascularity with age represent an intriguing aspect of meniscus tissue homeostasis and repair capacity that could contribute to an influence of age on PTOA. The main blood supply to the meniscus originates from branches of the geniculate arteries that ultimately penetrate the meniscus through its peripheral attachment to the surrounding capsular and synovial tissues. Branches of the geniculate arteries give rise to a perimeniscal capillary plexus that supplies the periphery, ranging from 10 to 30 % of the total meniscus width in humans [102]. The remaining central portion of the meniscus receives its nutrition by diffusion. The meniscus is fully vascular at birth, but during the second year of life, an avascular area develops in the inner region. By the second decade, the lateral third of the meniscus is vascularized followed by a decline to the outer quarter by age 50 years [103, 104]. Tears in the vascular (red zone) and vascular-avascular junction (red-white zone) can be repaired, while avascular tears (white zone) are typically resected [105]. Some clinicians have reported repairs in the avascular zone and have shown good outcomes in terms of initial improvement in symptoms, but higher rates of incomplete and failed healing present in these tears with age [106, 107].
Animal studies of meniscus healing have shown the expression of vascular endothelial growth factor (VEGF) at higher levels in the avascular region compared with the vascular region, and externally applied VEGF could not augment healing in avascular regions, suggesting that failure of healing in the avascular zone is not due to cellular inability to generate angiogenesis signals [108, 109], but rather due to other possible issues of intrinsic healing potential, vascular and cell repair supply, and growth or inhibitory factors.
Changes in Meniscus ECM
The function of the meniscus is reflected in the biologic and architectural composition of the extracellular matrix. Histologically, the meniscus is a fibrocartilaginous tissue populated primarily by fibrochondrocytes and type I collagen. Other extracellular matrix proteins include collagens II, III, V, and VI as well as elastin, proteoglycans, and glycoproteins [110, 111]. The extracellular matrix components are arranged to accommodate compressive, shear, and hoop stresses within the environment of the knee [112, 113]. It has been shown that meniscus tissue composition varies with the aging process, particularly in collagen fiber organization (Fig. 14.3).
Specific matrix proteins have been shown to be altered by aging in the meniscus. Perlecan, a large multidomain heparin sulfate- or chondroitin sulfate-substituted proteoglycan, declines with age relative to aggrecan and type I, II, and IV collagen [114]. Perlecan is present in the middle and inner meniscal zones and is expressed by cells of oval or rounded morphology [114]. In contrast to the other components visualized by this study which dropped marginally or remained relatively constant with age, perlecan was strongly cell associated, and its levels steadily declined with the onset of age and a loss of viable cells in the meniscus [114]. As an ECM component with roles in ECM organization, stabilization, cell attachment, and migration, and with binding capacity for fibroblast growth factors and connective tissue growth factor, the loss of perlecan with age has potential degenerative effects on meniscus ECM.
Meniscus total proteoglycan synthesis rates have been shown to be lower in older tissues (20–62 years) than in younger tissues (<20 years) with a higher proportion of decorin in younger tissues [115]. With age, there is an increase in aggrecan synthesis and mRNA expression as the major biosynthetic product of mature fibrochondrocytes. In contrast to lower synthesis rates with age, mRNA expression levels for decorin have been shown to increase with age [115]. Decorin is a small pericellular matrix proteoglycan that has the ability to bind type I collagen fibers and control fibril diameter thus contributing to its role in stabilizing and organizing the ECM of the meniscus [115]. Decorin also binds and sequesters the anabolic factor TGF-β which has a putative role in mediating intrinsic repair [115].
As a major structural component of the meniscus (60–70 % dry weight), the network of collagen fibers within the meniscus represents a major functional unit of the meniscus. Collagen cross-links occur via posttranslational modification in newly synthesized collagen, and intermolecular cross-links may play an important role in the pathologic progression of OA and aging in the meniscus. Pyridinoline and deoxypyridinoline are considered physiologic cross-links that maintain the structure of the collagen fibril and contribute to normal collagen function. The development of pathologic cross-links with age results in loss of elasticity of collagen and decreased proteolytic susceptibility upon accumulation of AGEs, such as pentosidine, which has been shown to increase with age [116, 117]. Analyses of OA and normal meniscus across a spectrum of ages have shown decreased pyridinoline and deoxypyridinoline cross-links with OA. In contrast, these cross-links did not change with age; rather, there was an exponential increase in the pathologic cross-link, pentosidine, with age consistent with susceptibility to age-associated degeneration of the meniscus. Both inner and outer regions of the meniscus demonstrated these findings [118].
Amyloid deposits can accumulate in certain tissues with age, and the meniscus seems to be particularly susceptible [119]. A study which examined the structure and chemical nature of amyloid in the meniscus determined that it was formed from deposits of apolipoprotein A-I made by meniscal cells [120]. Although the effects of amyloid deposits on the meniscus have not been determined, they could certainly interfere with normal meniscus function and cause cytotoxicity.
The biologic findings of reduced cellularity, diminished vascularity, and declining ECM properties are reflected in clinical findings. Although the available epidemiologic data may not be fully accurate, it is very uncommon for patients <10 years (high vascularity, cellularity, and healthy ECM) to present with acute meniscus tears unless they have a predisposing congenital abnormality such as a discoid meniscus [121]. With an extended vascular zone in such patients, attempted repair is almost always recommended regardless of tear morphology. In the adult and older population, healing is decreased in complex and degenerative tears as well as avascular tears [122, 123]. With age (>40 years), more tears have complex and horizontal morphology compared with bucket-handle, longitudinal, and radial tears being dominant in young as well as more tears occurring in the medial compartment in old versus young [122]. Age alone is not a contraindication to repair, but repair is less likely to be pursued due to degenerative tissue quality and tear complexity rather than exclusion with age alone [122, 123].
While these studies collectively provide important insight into the role that the meniscus plays in the biology of age-associated PTOA, they are merely a starting point for truly understanding this process. With the incidence of degenerative meniscus tears increasing with advancing age, gaining a better understanding of the mechanisms of meniscus degeneration could lead to better therapeutic strategies and interventions for meniscus disorders and OA prevention.
Ligaments
Joint injury is often associated with damage to ligaments, and in particular in the knee joint, ACL rupture is a major determinant of the risk for the development of PTOA [124, 125]. The ACL is essential for knee kinematics especially in rotation and functions as an anterior/posterior stabilizer [126, 127]. In the setting of ACL deficiency, the articular cartilage, as well as the menisci, in the medial tibiofemoral compartment is more susceptible to arthritic change than the lateral compartment [128]. A large number of OA patients without prior history of ligament injury have ACL deficiency at the time of total knee arthroplasty [10, 129], and a correlation between the radiologic OA grade and the histological grade of ACL degeneration has been reported in end-stage OA [130]. In addition, ACL rupture is more common among patients with symptomatic knee OA. As noted above, between 22 and 35 % of these patients have incidental complete ACL tears identified by MRI [10, 131, 132]. It has been reported that fewer than half of subjects with ACL rupture recall a knee injury, suggesting that this risk factor for knee OA is under recognized [10].
Histological abnormalities in ACL are highly prevalent in knees with severe OA and include cystic changes, disorientation of collagen fibers, and mucoid degeneration [130]. However, histological changes are present at early stages and can precede cartilage histopathology. An analysis of the ACL in a large number of human knee joints across the entire adult age spectrum addressed aging-related changes in the ACL and their relationship with changes in cartilage [133]. Degenerated ACL was found in knees without cartilage degeneration. Also, knees with minimal cartilage degeneration can have moderate to severe ACL damage. These findings suggest that ACL degeneration can be initiated before or progresses more rapidly than cartilage degeneration, at least in a subpopulation of individuals [133].
Changes in Ligament ECM
The earliest and most prevalent age-related change in the ACL extracellular matrix is disorganization of collagen fibers, which can be seen in ACL from young donors without cartilage degeneration or inflammation (Fig. 14.4) [133]. An aging-related decrease in the diameter of the collagen fibrils and a corresponding increase in the concentration of small fibrils have been described for the human ACL [134]. Mucoid degeneration of ligaments refers to the presence of discontinuous and disorganized collagen fibers and the presence of stainable mucoid matrix (Fig. 14.4) [135, 136]. Histologically, mucoid degeneration is highly prevalent and can be observed in ACL from young donors with normal cartilage [133]. Cystic changes represent areas within fascicles that are devoid of extracellular matrix and are a relatively late event in ACL degeneration [130, 137, 138]. Calcium deposition within ACL is evident as slightly basophilic material, compatible with calcium pyrophosphate dihydrate crystals [139]. Crystal deposition is reported to occur with a frequency of 0.1 % of adult persons and to increase with age and is typically seen in older donors (age >70 years) that also have degenerated cartilage [133].
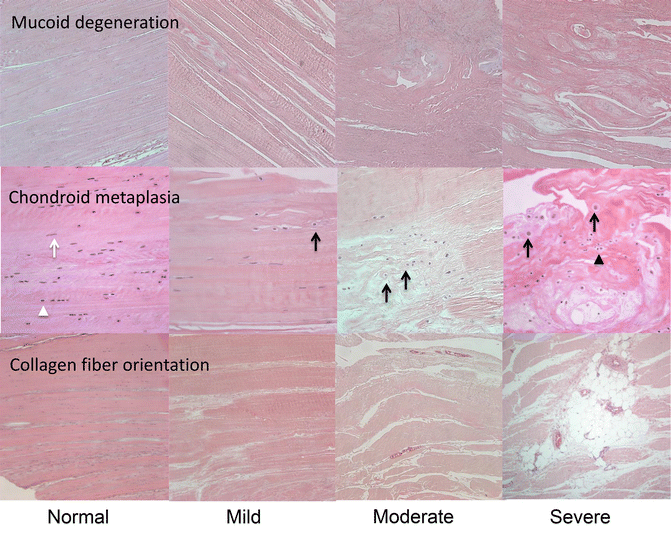
Fig. 14.4
Aging-associated changes in ACL. ACLs were collected from human knees at autopsy. Representative samples were selected to illustrate different types and severities of aging-associated changes
MMP-3 is a key enzyme involved in extracellular matrix degeneration [140]. Synovial fluid MMP-3 concentrations were markedly higher in knees with ruptured ACL than in normal knees [141]. MMP-3 may originate from synovial and inflammatory cells but also from cells within the ACL itself. Even normal human ACL expresses MMPs [142]. The average percentage of MMP-3 positive cells in ACL from normal knees decreased with aging, but increased in ACL from knees with severe cartilage degeneration. Cells expressing MMP-3 in degenerated ACL were predominantly cell aggregates of chondrocyte-like cells but not fibroblast-like cells. These results suggest that the decrease of MMP-3 positive cells and total cell number density with aging may reflect a reduced capacity to remodel and maintain the tissue, while increased MMP-3 positive cells in ACL from knees with severe cartilage degeneration may be caused of phenotypic changes and contribute to degeneration.
ACL Cellular Changes
Major changes in cell density, organization, and phenotype occur during ACL aging and in OA-affected joints. First, there appears to be a general reduction in cell density with age in joints that do not have cartilage destruction. Aging-related changes in ligament cell response to growth factors may contribute to cell loss [143]. This cell loss is seen uniformly in all regions of the ACL and within all tissue layers. The reduction in cellularity may compromise the ability of the ACL to maintain homeostasis and respond to injury [144]. At a certain point following cell depletion or damage to the ECM, focal areas with increased cell density emerge. This occurs in two distinct patterns, one where fibroblastoid cells are found concentrated around blood vessels and a second consisting of patches with high numbers of chondrocyte-like cells. This local increase in cell numbers may be the result of cell proliferation, and in the perivascular areas, it is possible that fibroblast-like cells are infiltrating from the circulation.
Related posts:
 Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
Arthritis That Develops After Joint Injury: Is It Post-Traumatic Arthritis or Post-Traumatic Osteoarthritis?
Post-Traumatic Arthritis: Definitions and Burden of Disease
Potential Targets for Pharmacologic Therapies for Prevention of PTA
 Outcomes of ACL Injury: The MOON Consortium
Outcomes of ACL Injury: The MOON Consortium
 Biomarkers of PTA
Biomarkers of PTA
 Instability: Dynamic Loading Models
Instability: Dynamic Loading Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


