Chapter 21
Acid-Base Disorders (Case 14)
Kavita Ahuja DO and Ilene Miller MD
Case: A 46-year-old man is found unconscious on the bathroom floor by his family. The family states the patient has a medical history significant only for depression and gastroesophageal reflux disease (GERD). His medications include escitalopram and omeprazole. Upon arrival to the emergency department the patient is more responsive but still confused and drowsy. A few episodes of vomiting are noted. On exam, the patient is afebrile with a blood pressure (BP) of 90/50 mm Hg, respiratory rate (RR) of 24 breaths per minute, and heart rate (HR) of 120 beats per minute (bpm). His pupils are reactive to light with a nonfocal neurologic exam. He has dry mucous membranes and mild diffuse abdominal tenderness to palpation. No alcoholic fetor is noted. Laboratory studies are as follows: Na 132 mEq/L, K 3.4 mEq/L, Cl 90 mEq/L, serum bicarbonate (HCO3) 10 mEq/L, BUN 60 mg/dL, Cr 1.6 mg/dL, and glucose 80 mg/dL. Arterial blood gas (ABG) reveals a pH of 7.2 and PCO2 of 25 mm Hg. Serum ethanol, acetone, and serum β-hydroxybutyrate are negative. The measured serum lactate is 1.6 mmol/L. Urinalysis reveals 4+ calcium oxalate crystals.
Differential Diagnosis
Increased Anion Gap Metabolic Acidosis | Normal Anion Gap Metabolic Acidosis | Metabolic Alkalosis |
Methanol and ethylene glycol poisoning | Gastrointestinal (GI) bicarbonate loss | Contraction alkalosis |
Ketoacidosis | RTA | Milk alkali syndrome |
Lactic acidosis | ||
Uremia | ||
Salicylate intoxication |
Speaking Intelligently
When encountering a patient whose workup reveals a significant acid-base disorder, a thorough history and physical exam, along with appropriate lab data, will help identify the underlying diagnosis. These patients may require immediate attention (i.e., hemodynamic stabilization, volume resuscitation, and optimization of oxygenation). An ABG is the best test to assess the presence of an acid-base disorder. If a toxic ingestion is suspected, a toxicology screen should be performed to aid in identifying the agent, and a poison control center should be contacted for additional guidance.
PATIENT CARE
Clinical Thinking
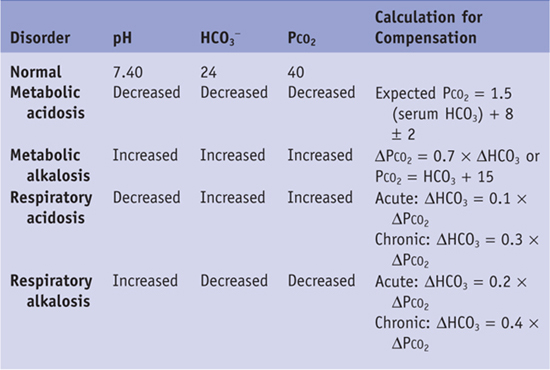
• Metabolic acidosis is associated with a low pH and low HCO3 concentration.
• Metabolic alkalosis consists of a high pH with an increased HCO3 concentration.
• Respiratory alkalosis is associated with a high pH and a low PCO2.
• The approach to identifying and managing acid-base disturbances should be a stepwise approach, as follows (also see Table 21-1):
4. Next, calculate the anion gap to aid in your differential diagnosis. It is helpful to classify the metabolic acidosis into increased anion gap versus normal anion gap. The anion gap is the difference between measured serum cations and anions and can be calculated as Na+ − (Cl− + HCO3−). A normal anion gap is 12 ± 4. An increase in anion gap is most often caused by an increase in unmeasured anions, such as lactate or ketones. The anion gap in this patient is 32. Therefore, this patient has a high–anion gap metabolic acidosis with appropriate respiratory compensation.
Table 21-1 Expected Changes in Primary Acid-Base Disorders
History
Physical Examination
• Determine if the patient is altering his or her respiratory rate in an attempt to compensate for alterations in serum HCO3.
• Look for signs of inebriation.
• Look for signs of malnutrition, as they may raise suspicion of alcoholic ketoacidosis.
Tests for Consideration
| Clinical Entities | Medical Knowledge |
Methanol and Ethylene Glycol Poisoning | |
Pφ | Two toxic alcohol ingestions that lead to a severe anion-gap acidosis are methanol and ethylene glycol. The low molecular weight of these substances allows for entry into cells. Methanol and ethylene glycol are metabolized to formic acid and glycolic acid, respectively, and this metabolism generates H+ ions, resulting in a high–anion gap acidosis. These substances are found in automotive antifreeze and de-icing solutions, windshield wiper fluid, solvents, and cleaners, and are ingested accidentally or as a suicide attempt. |
TP | Patients with methanol ingestion present with GI symptoms (abdominal pain, vomiting), central nervous system (CNS) symptoms (inebriation, headache, lethargy, coma), and an anion gap metabolic acidosis usually 12–24 hours after ingestion. No alcoholic fetor is present. An additional clue to the diagnosis of methanol ingestion is the presence of visual disturbances, as formic acid causes retinal injury. Ethylene glycol is found in antifreeze, and its ingestion has a similar clinical presentation but does not produce visual disturbances. The toxic metabolite of ethylene glycol causes kidney damage, and these patients often present with renal failure. Ethylene glycol, in addition to being metabolized to glycolic acid, is metabolized to oxalate, causing precipitation of calcium oxalate crystals, which can cause flank pain and further deterioration in renal function. Calcium oxalate crystals can be detected in the urine and may be seen in the form of calcium oxalate dihydrate (envelope-shaped crystals) or, more commonly, calcium oxalate monohydrate (needle-shaped or dumbbell-shaped crystals). Additionally, the urine will be fluorescent under a Wood lamp. |
Recognition of these ingestions should be made early, as prompt therapy is associated with significant decrease in morbidity and mortality. An increased osmolar gap is an early clue to diagnosis. The osmolar gap is defined as the difference between the measured serum osmolality and the calculated osmolality. An osmolar gap >15 mOsm/kg indicates that a toxic ingestion is the likely diagnosis. Of note, an osmolar gap can also be present in other conditions that are not associated with a high–anion gap metabolic acidosis, such as ethanol or isopropyl alcohol ingestion. Ethylene glycol poisoning can also cause hypocalcemia. | |
Tx | Because the toxicity of methanol and ethylene glycol is due to the metabolites, therapy is directed at inhibiting metabolism of these substances. Fomepizole is an agent that blocks the metabolism of these agents by inhibiting alcohol dehydrogenase. Intravenous ethanol has also been used to competitively inhibit alcohol dehydrogenase and prevent formation of glycolic acid. Sodium bicarbonate can be used to correct acidosis. Hemodialysis, which allows for removal of toxic metabolites, is another modality that can be instituted if severe metabolic derangement or end-organ damage is present. See Cecil Essentials 28. |
Lactic Acidosis | |
Pφ | Lactic acidosis is a frequent cause of increased anion gap in hospitalized patients. Lactate is the end product of anaerobic metabolism of glucose, and, under normal conditions, its production is minimal. When lactate production increases faster than lactate can be metabolized, lactic acidosis ensues. Lactic acid exists in two forms, called isomers: L-lactate and D-lactate. L-lactate is produced in human metabolism and increases with impairments in tissue oxygenation secondary to increased anaerobic metabolism. D-lactate is produced by bacteria and is therefore primarily seen in patients with colonic bacterial overgrowth such as those with short-gut syndrome, gastric bypass, or small bowel resection. |
Patients with lactic acidosis can present with abdominal pain, nausea, vomiting, CNS symptoms, and alterations in respiration. The severity of symptoms varies with the cause and degree of acidosis. L-lactic acidosis can be classified as type A or type B lactic acidosis. Type A lactic acidosis occurs in the setting of tissue hypoperfusion and is usually seen in shock, cardiopulmonary arrest, and/or hypoxemia. A transient lactic acidosis may be seen with seizures or excessive exercise. Type B lactic acidosis is found in patients without symptoms of hypoperfusion and often results from medications that interfere with lactate or pyruvate metabolism, such as metformin or reverse transcriptase inhibitors. | |
Dx | The diagnosis of lactic acidosis should be considered in all cases of anion gap metabolic acidosis. In lactic acidosis, the anion lactate accounts for the increase in anion gap. Lactic acidosis is diagnosed when elevated levels of serum lactate are demonstrated. Routine serum lactate assays measure the L-lactate isomer but do not detect the D-lactate isomer. If D-lactic acidosis is suspected, the appropriate assay should be ordered. |
Tx | Treatment of lactic acidosis is aimed at treatment of the underlying disorder. For example, in septic shock, treatment should include restoring adequate tissue perfusion with the use of IV fluids and vasopressor support. The role of HCO3 in lactic acidosis is controversial, as HCO3 therapy may worsen intracellular acidosis. See Cecil Essentials 28. |
Ketoacidosis | |
Pφ | Ketones are produced when the body metabolizes adipose tissue for energy and the liberated fatty acids are metabolized to ketoacids (β-hydroxybutyric and acetoacetic acid), which can be detected in serum and urine. The two most common types of ketoacidosis are diabetic ketoacidosis (DKA) and alcoholic ketoacidosis (AKA). In both situations a decrease in the insulin-to-glucagon ratio leads to fatty acid mobilization and ketoacid accumulation. |
Patients with DKA typically present with hyperglycemia, polyuria, polydipsia, nausea, vomiting, abdominal pain, and hyperventilation. Physical exam may reveal signs of volume depletion resulting from a combination of an osmotic diuresis from hyperglycemia along with vomiting and poor oral intake. Fruity odor and hyperventilation may also be noted. AKA should be suspected in a patient with a history of alcohol abuse with recent binge drinking and poor oral intake. These patients, in contrast to those with DKA, may be hypoglycemic on presentation. | |
Dx | The diagnosis of DKA is made in a hyperglycemic patient with a high–anion gap acidosis and detection of serum or urine ketones. AKA is diagnosed in a patient with a history of alcohol abuse and high–anion gap acidosis with positive serum or urine ketones. The predominant ketone in AKA is β-hydroxybutyrate. |
Tx | Therapy for DKA consists of administration of insulin, as well as volume and electrolyte repletion. Close monitoring of glucose levels and serum chemistries is essential. The treatment of AKA similarly consists of volume resuscitation and electrolyte repletion. Additionally, these patients require glucose and thiamine administration. Glucose administration stimulates insulin release and stops fatty acid generation. See Cecil Essentials 28, 69. |
Uremia | |
Pφ | Early in the course of renal failure, a non–anion gap, hyperchloremic metabolic acidosis may be present due to impaired ammonium excretion. However, as the GFR decreases further, the kidney becomes unable to excrete organic and inorganic anions, causing a high–anion gap metabolic acidosis. |
TP | Patients generally present with symptoms secondary to renal failure and the resulting metabolic acidosis. These symptoms include anorexia, nausea, vomiting, hyperventilation, congestive heart failure, muscle weakness, and changes in mental status. |
Dx | The presence of an elevated BUN and creatinine with acidosis suggests that renal failure is the cause of metabolic acidosis. |
Tx | Treatment of metabolic acidosis secondary to uremia consists of HCO3 supplementation to restore normal blood pH and alleviate symptoms. Diuretics are often used in cases of volume overload. Refractory cases of metabolic acidosis may warrant initiation of dialysis. See Cecil Essentials 33. |
Pφ | Salicylate (aspirin) intoxication is another cause of elevated anion gap metabolic acidosis. At toxic concentrations, salicylate uncouples oxidative phosphorylation and leads to increased production of organic acids. Aspirin is widely available, and therefore salicylate intoxication is a common poisoning. |
TP | The symptoms of salicylate intoxication are dose-dependent, with most patients showing signs of toxicity at plasma concentrations >40–50 mg/dL. Common clinical manifestations include nausea, vomiting, tinnitus, pulmonary edema, altered mental status, and coma. Adults may present with a mixed disorder of high–anion gap acidosis with respiratory alkalosis, as salicylate stimulates the respiratory center, causing increased ventilation. Severe intoxication can lead to death from CNS toxicity and cardiorespiratory depression. |
Dx | The diagnosis is made in the setting of an increased anion gap acidosis in combination with a respiratory alkalosis in a patient with an elevated plasma salicylate concentration. |
Tx | Treatment includes GI decontamination with activated charcoal, correction of fluid and electrolyte disorders, and facilitating excretion of the drug. HCO3 infusion will cause alkalemia and alkalinization of the urine and therefore inhibit diffusion of salicylate into brain tissue while promoting renal excretion. Hemodialysis should be considered in patients with extremely high salicylate levels (usually >100 mg/dL), severe neurologic disturbances, renal failure, or refractory acidosis. See Cecil Essentials 28. |
GI Loss of HCO3 | |
Pφ | Intestinal fluid distal to the stomach has a high concentration of HCO3, and GI HCO3 loss is a common cause of metabolic acidosis. The HCO3 loss most commonly occurs with diarrhea but can also be seen with excessive biliary and/or pancreatic fluid loss. The excessive volume loss causes the kidneys to reabsorb NaCl, resulting in a normal anion gap hyperchloremic acidosis. The kidneys, in response to acidosis, will increase urinary excretion of NH4+, which will lead to a negative urine anion gap. |
TP | Patients typically present with a history of diarrhea and serum chemistries showing both a normal anion gap acidosis and hypokalemia. With excessive volume loss, they can present with hypotension and tachycardia. |
The diagnosis of GI HCO3 loss as the etiology of metabolic acidosis is made by detection of a normal anion gap metabolic acidosis with a negative urine anion gap and a clinical history suggestive of GI HCO3 loss. | |
Tx | Treatment is aimed at treating the cause of GI HCO3 loss. Those with severe acidosis or prolonged acidosis require HCO3 repletion. Hypokalemic patients require potassium repletion. See Cecil Essentials 28. |
Renal Tubular Acidosis | |
Pφ | RTA refers to a group of disorders characterized by hyperchloremic, normal anion gap metabolic acidosis as a result of impaired renal HCO3 absorption or H+ secretion in the setting of a normal GFR. RTA may be a primary disorder or secondary to a number of systemic diseases such as diabetes mellitus, multiple myeloma, or autoimmune diseases. Medications and toxins have also been known to cause RTA. |
TP | Patients are generally asymptomatic but may present with nonspecific symptoms related to the underlying cause of RTA. Patients with type I RTA often develop nephrolithiasis. Patients with type IV RTA are often hyperkalemic. However, often RTA may be incidentally discovered on routine blood screening. |
Dx | RTA is suspected in the presence of a normal anion gap, hyperchloremic metabolic acidosis in conjunction with a positive urine anion gap. This indicates an inability of the kidney to excrete NH4. |
Tx | Treatment of RTA includes identifying the underlying cause along with administration of alkali (i.e., sodium bicarbonate) to correct the acidosis. Potassium supplementation is often necessary. Thiazide diuretics may also be used in some instances to increase proximal tubule HCO3 reabsorption. See Cecil Essentials 28. |
Contraction Alkalosis | |
Pφ | Contraction alkalosis is a term used to describe a combined presentation of volume contraction and primary metabolic alkalosis. It is seen in patients with vomiting and diuretic use and results from the loss of HCO3-free fluid. The alkalosis is then maintained by chloride depletion. Specific cells in the distal tubule exchange chloride for HCO3, and thus chloride depletion will not allow HCO3 excretion and correction of the alkalosis. |
Metabolic alkalosis is generally well tolerated and is only discovered on routine laboratory testing. However, patients with serum HCO3 concentrations > 50 mmol/L may manifest with neurologic symptoms such as seizures, delirium, or stupor. Other symptoms may be related to other associated abnormalities such as hypokalemia and volume depletion. | |
Dx | The diagnosis requires a history suggesting volume depletion and Cl− loss such as vomiting, nasogastric suctioning, and/or diuretic use. Laboratory data reveal increased serum HCO3, hypokalemia, and an elevated arterial pH. Urine electrolytes show a urine Cl− < 20 mmol/L. |
Tx | Treatment involves correction of the underlying disorder and volume expansion with NaCl. The distal tubular cells exchange HCO3 for Cl− and correct the alkalosis. Electrolytes such as K+ and Mg2+ may need to be supplemented. See Cecil Essentials 28. |
Milk-Alkali Syndrome | |
Pφ | Milk-alkali syndrome is characterized by metabolic alkalosis, hypercalcemia, and renal insufficiency. It occurs in the setting of continuous alkali (sodium bicarbonate) ingestion along with excess calcium intake (milk or CaCO3). The hypercalcemia produces a reduction in GFR. This reduction in GFR does not allow the kidneys to excrete HCO3, and metabolic alkalosis develops. |
TP | Approximately 50% of these patients are asymptomatic. The remaining 50% present with symptomatic hypercalcemia, including nausea, vomiting, weakness, muscle aches, polyuria, polydipsia, nephrolithiasis, and mental status changes. |
Dx | A history of calcium and alkali ingestion (often in the form of calcium carbonate), along with laboratory data indicating hypercalcemia and metabolic alkalosis, suggests milk-alkali syndrome. |
Tx | Treatment consists of withdrawal of the offending agent, which generally leads to rapid improvement. Symptomatic patients can be treated with isotonic saline and loop diuretics. Renal insufficiency is reversible and improves with correction of hypercalcemia. See Cecil Essentials 28, 74. |





