Molecular diagnosis is an important aspect in the care of patients with neuromuscular disorders. Because of the rapidly evolving nature of the field, the approach to obtaining a molecular diagnosis may be challenging. This article provides a general approach to molecular diagnostic testing while reviewing the principles of genetics and genetic disorders and the indications and limitations of testing methods in common hereditary neuromuscular disorders.
- •
Molecular testing is an important aspect in the care of patients with neuromuscular disorders.
- •
Some potential benefits include diagnostic certainty, the identification of current and future therapeutics, and information to help guide accurate genetic counseling and family planning.
- •
Once a genetic condition is considered, it is usually imperative to establish as precise a clinical diagnosis as possible before proceeding with targeted molecular diagnostic testing.
- •
The future of molecular testing will likely shift away from targeted genetic analysis toward genomic technologies to allow for a nontargeted approach.
Introduction
Genetic testing plays an increasingly important role in neuromuscular medicine. Over the last 2 decades, there has been rapid progress in molecular genetics, leading to a continually increasing number of identified diagnostic entities and clinically available diagnostic tests. As genetic testing becomes more efficient from a cost and throughput standpoint, it will become an increasingly useful part of patient care. This practicality is particularly likely as treatments become available; as our understanding of hereditary neuromuscular disorders increases and potential treatment strategies are identified, genetic testing will become critical to provide personalized care. For the clinician, this growth in genetic testing may be daunting, and the approach to obtaining a molecular diagnosis may seem challenging.
The utility of genetic testing is often questioned by physicians, patients, or insurers when specific treatments are unavailable for most of the known hereditary conditions. However, the accepted benefits of genetic testing extend beyond treatment alone. Perhaps most importantly, genetic testing can often provide a definitive diagnosis, shortening the diagnostic odyssey for a family and avoiding unnecessary and often expensive additional testing; provide information for accurate genetic counseling and family planning guidance; suggest appropriate surveillance testing; and identify current and future therapeutic interventions. Benefits beyond standard clinical care include the identification of specific mutations, potentially allowing clinical trial participation, or participation in genotype/phenotype correlation and natural history studies.
These benefits may come at a significant financial burden, and striking a balance between sufficient and excessive use of testing is not always easy or straightforward. Many patients require multiple tests to achieve a diagnosis, and testing may exceed thousands of dollars. In some cases, despite extensive testing, a specific mutation may not be identified. In such cases, the possibility of an underlying hereditary condition may not be entirely excluded because of the incomplete understanding of the hereditary causes of most neuromuscular disorders. Additional potential drawbacks of genetic testing include psychological stress and anxiety and potential stigmatization and discrimination with the confirmation of a genetic disorder. The identification of a genetic disorder in presymptomatic patients may cause unnecessary psychological stress. Additionally, genetic testing in an affected individual carries significant potential implications for family members at risk of carrier or affected status.
For these reasons, the clinician involved in the care of neuromuscular disease needs a working understanding of the principles of genetics and genetic diagnoses as well as the indications for performing and the limitations of common genetic tests. Furthermore, they must be prepared to provide genetic testing results along with (or with access to) appropriate genetic counseling.
Hereditary basis of disease
For most disorders related to single gene inheritance, disease phenotype is caused by the inheritance of a mutated copy or copies of a gene. The pattern of inheritance is determined by whether the mutated gene is on an autosome (chromosomes 1–22) or on a sex-linked chromosome (chromosome X or Y) and whether the disorder is dominant or recessive ( Fig. 1 ). Autosomal disorders affect both sexes and can be transmitted by either sex. Individuals with autosomal dominant disorders usually have at least one affected parent, and subsequent generations of an affected parent have a 50% chance of being affected. A gene is considered dominant if one copy is sufficient to determine the presence of a particular trait. Autosomal recessive disorders are caused by having 2 mutated copies of a gene, one copy from each parent. Parents of an affected individual are usually asymptomatic but carry one mutated copy of a gene. Children of 2 asymptomatic carriers have a 25% chance of being affected.
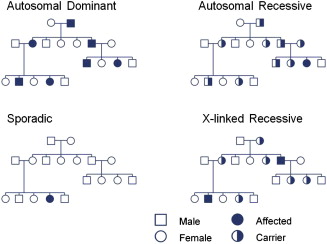
X-linked disorders may occur in a dominant or recessive fashion. Females have 2 X chromosomes, one of paternal origin and the other of maternal origin. Males have one Y chromosome of paternal origin and one X chromosome of maternal origin. X-linked recessive disorders usually only affect males born to asymptomatic parents. The father is unaffected and (usually) the mother is an asymptomatic carrier. In X-linked disorders, there is no male-to-male transmission. Because of lyonization, or inactivation of one X chromosome in females, a proportion of females may be symptomatic carriers of recessive X-linked traits and this can occur in 2% to 8% in Duchenne muscular dystrophy (DMD).
Mitochondrial inheritance (not shown in see Fig. 1 ) is another important mode of inheritance. Mitochondrial DNA is transmitted from maternal origin because the mitochondria from the sperm are usually actively degraded after fertilization. The transmission occurs from a maternal lineage, and all offspring may be affected. Mitochondrial DNA mutations may subsequently be transmitted from female but not male offspring to future generations. However, it is important to note that not all mitochondrial disorders are caused by mitochondrial DNA mutations and may also be related to mutations in autosomally encoded mitochondrial proteins.
In addition to the previously discussed modes of transmission, sporadic or de novo mutations may spontaneously occur. In this instance, family history or genetic testing of parents for affected or carrier status is unrevealing. This phenomenon occurs with variable frequency and occasionally may exceed the frequency of inherited mutations in certain conditions. When family history is negative, a de novo mutation should be considered, but other possible explanations exist. Somatic mosaicism may occur in which a mutation may be in some but not all cells; if the mutation is in germline cells, it may be passed onto future generations. Other issues, including age of onset, mistaken paternity, and incomplete penetrance, can obscure a significant family history and should be taken in account when obtaining and reviewing the family history.
Hereditary basis of disease
For most disorders related to single gene inheritance, disease phenotype is caused by the inheritance of a mutated copy or copies of a gene. The pattern of inheritance is determined by whether the mutated gene is on an autosome (chromosomes 1–22) or on a sex-linked chromosome (chromosome X or Y) and whether the disorder is dominant or recessive ( Fig. 1 ). Autosomal disorders affect both sexes and can be transmitted by either sex. Individuals with autosomal dominant disorders usually have at least one affected parent, and subsequent generations of an affected parent have a 50% chance of being affected. A gene is considered dominant if one copy is sufficient to determine the presence of a particular trait. Autosomal recessive disorders are caused by having 2 mutated copies of a gene, one copy from each parent. Parents of an affected individual are usually asymptomatic but carry one mutated copy of a gene. Children of 2 asymptomatic carriers have a 25% chance of being affected.
X-linked disorders may occur in a dominant or recessive fashion. Females have 2 X chromosomes, one of paternal origin and the other of maternal origin. Males have one Y chromosome of paternal origin and one X chromosome of maternal origin. X-linked recessive disorders usually only affect males born to asymptomatic parents. The father is unaffected and (usually) the mother is an asymptomatic carrier. In X-linked disorders, there is no male-to-male transmission. Because of lyonization, or inactivation of one X chromosome in females, a proportion of females may be symptomatic carriers of recessive X-linked traits and this can occur in 2% to 8% in Duchenne muscular dystrophy (DMD).
Mitochondrial inheritance (not shown in see Fig. 1 ) is another important mode of inheritance. Mitochondrial DNA is transmitted from maternal origin because the mitochondria from the sperm are usually actively degraded after fertilization. The transmission occurs from a maternal lineage, and all offspring may be affected. Mitochondrial DNA mutations may subsequently be transmitted from female but not male offspring to future generations. However, it is important to note that not all mitochondrial disorders are caused by mitochondrial DNA mutations and may also be related to mutations in autosomally encoded mitochondrial proteins.
In addition to the previously discussed modes of transmission, sporadic or de novo mutations may spontaneously occur. In this instance, family history or genetic testing of parents for affected or carrier status is unrevealing. This phenomenon occurs with variable frequency and occasionally may exceed the frequency of inherited mutations in certain conditions. When family history is negative, a de novo mutation should be considered, but other possible explanations exist. Somatic mosaicism may occur in which a mutation may be in some but not all cells; if the mutation is in germline cells, it may be passed onto future generations. Other issues, including age of onset, mistaken paternity, and incomplete penetrance, can obscure a significant family history and should be taken in account when obtaining and reviewing the family history.
General approach
Because of the significant costs of most molecular testing, it is imperative to establish as precise a clinical diagnosis as possible. The most important and most obvious step is the consideration that patients’ symptoms may be genetic in origin. The possibility of an inherited process is not always obvious. The presence of slowly progressive or longstanding symptoms and other features listed in Box 1 may suggest a hereditary process but such features are occasionally absent. It is not uncommon for patients to be initially unable to identify slowly progressive deficits or recognize similar symptoms in other family members, particularly if other family members have not received a confirmed diagnosis. Slowly progressive symptoms often go unnoticed until such symptoms interfere with day-to-day activities. Specific questions regarding early milestones, participation in sports, or other physically demanding activities is often necessary to reveal subtle deficits in neuromuscular function. The examination of other unaffected family members may be required to determine subtle involvement.
Longstanding or slowly progressive deficits
Clinical signs out of proportion to the patients’ described symptoms
History of similar symptoms in other family members
Early onset
Musculoskeletal abnormalities, such as pes cavus, scoliosis, contractures
If a genetic cause is considered, the general approach begins with clinical localization and use of ancillary testing as necessary to determine as precise a diagnosis as possible before obtaining molecular genetic testing. Depending on the clinical phenotype, electrodiagnostic testing, laboratory testing, or tissue biopsy may be required to exclude other acquired disorders and narrow the differential diagnosis to allow for targeted molecular testing. Despite these measures, the diagnostic yield of neurogenetic testing can be low even if multiple tests are pursued. The combined yield in a retrospective study was as low as 20%.
All patients who are at risk to be affected by genetic disorders should be offered genetic counseling. Inheritance patterns, risk to offspring, and potential implications for other family members should be clearly explained. With this information, families can make informed decisions to help deal with risks and potentially lessen the impact of the disease. In some cases, the information can assist in making financial, lifestyle, family planning, and career choices.
This article focuses on general patterns of hereditary neuromuscular disorders ( Table 1 ) and the common hereditary conditions within each subtype. The disorders are organized by localization (nerve, muscle, motor neuron, and neuromuscular junction), and the clinical features characteristic for individual disorders are outlined. Because of the constraints of the article, not all hereditary conditions with peripheral nervous system involvement are discussed (ie, hereditary spastic paraparesis, spinocerebellar ataxia, Friedreich ataxia, porphyria, and so forth).
| Patterns of Hereditary Neuromuscular Disorders | Common Features |
|---|---|
| Myopathies | |
| Muscular dystrophy | Usually proximal predominant weakness |
| Distal myopathy | Distal muscle weakness |
| Congenital myopathy | Early onset with static or slowly progressive weakness |
| Metabolic myopathy | Slowly progressive or dynamic symptoms of weakness, ± exertional rhabdomyolysis and muscle contractures |
| Mitochondrial myopathies | Weakness ± other systemic features of mitochondrial cytopathy |
| Muscle channelopathy | Fluctuating symptoms of muscle stiffness (myotonia) or weakness (periodic paralysis) |
| Motor Neuron | |
| Proximal spinal muscular atrophy | Lower motor neuron dysfunction with proximal muscle weakness |
| Familial ALS | Mixed lower and upper motor neuron dysfunction |
| Distal spinal muscular atrophy | Distal muscle weakness |
| Spinobulbar muscular atrophy | Lower motor neuron dysfunction with proximal limb and bulbar weakness |
| Neuropathy | |
| Charcot-Marie-Tooth | Distal sensory loss and muscle weakness |
| Hereditary sensory and autonomic neuropathy | Sensory loss Variable autonomic dysfunction Usually less prominent weakness |
| Distal hereditary motor neuropathy | Distal weakness Occasional mild sensory loss |
| Focal/multifocal neuropathy | Recurrent bouts of focal sensory loss and weakness |
| Nerve channelopathy | Generalized neuropathy with variable features of sensory loss or pain |
| NMJ disorders | |
| Congenital myasthenic syndromes | Fluctuating weakness |
Hereditary neuropathies
Charcot-Marie-Tooth Disease
Charcot-Marie-Tooth (CMT) disease is a group of hereditary disorders with significant clinical and genetic heterogeneity characterized by variably present findings of slowly progressive distal muscle weakness and wasting, sensory loss, and reduced reflexes. The prevalence of CMT is approximately 1 in 2500. Rapid advancement in genetic understanding has brought both clarification and confusion into classification strategies because of genotypic and phenotypic variability; despite significant advances, diagnostic strategies are limited by the incomplete knowledge of mutations associated with CMT. Up to one-third of patients with clinically defined CMT will have genetically undefined disorders even if all known mutations are assessed. The classification of CMT subtypes is based on electrophysiological criteria of axonal loss versus demyelination and inheritance pattern. Electrophysiology is usually stratified into demyelinating, intermediate, and axonal subtypes; diagnostic algorithms are available elsewhere to help guide genetic testing. Demyelinating subtypes (autosomal dominant [CMT1], autosomal recessive [CMT4], and X-linked recessive [CMTX]) are characterized by electrophysiological findings of upper limb motor conduction velocities less than 38 m/s. Axonal subtypes (CMT2) are characterized by motor conduction velocities greater than 45 m/s, and intermediate subtypes are characterized by motor conduction velocities between greater than 35 m/s and greater than or equal to 45 m/s.
Nerve conduction studies are thus the starting point for the evaluation of patients with CMT and are necessary to guide rationale testing. CMT1A is the most common form of all CMT and is responsible for close to 90% of patients with demyelinating CMT with motor conduction velocities between 15 and 35 m/s. It is inherited in an autosomal dominant pattern, but 10% of cases may occur related to a de novo mutation. It is caused by a 1.5 Mb duplication of the PMP22 gene on chromosome 17p11.2. In cases with intermediate upper limb motor nerve conduction velocities, CMTX (caused by mutations in the GJB1 gene) and CMT1B ( MPZ gene) are responsible for approximately 50% and 25% respectively. Axonal forms (CMT2) make up only around 12% of CMT cases. Among them, CMT2A2 (caused by mutations in the MFN2 gene) is the most common cause at 20%. Besides these specific genes, however, all other causes are exceedingly rare, making shotgun testing of all CMT genes inefficient.
Hereditary sensory and autonomic neuropathy (HSAN) is a related but distinct group of disorders associated with progressive sensory loss, variable autonomic disturbance, and less frequent/prominent loss of motor function. HSAN is significantly less common than hereditary motor and sensory neuropathy. Usually, this group of disorders is classified by clinical characteristics, inheritance pattern, and electrodiagnostic findings.
Distal Hereditary Motor Neuropathies
The distal hereditary motor neuropathies (dHMN), also referred to as distal spinal muscular atrophies and sometimes classified within motor neuron disorders, are an unusual group of disorders characterized by distal weakness and muscle atrophy. Electrodiagnostic features usually show distal motor axon loss with preserved conduction velocities and normal sensory responses. dHMN is classified clinically by inheritance and associated clinical features, such as age of onset; inheritance pattern; and other distinguishing features, such as vocal cord paralysis, upper limb predominance, or upper motor neuron signs. Several genes have been identified that are associated with the clinical phenotype of dHMN. The symptoms of dHMN overlap significantly with that of CMT and are distinguished only by preserved sensation, but it is important to note that sensory loss is not invariably absent. This clinical heterogeneity is reflected in the case of mutations in HSPB1 , which has been associated with both dHMN and CMT (CMT2F) phenotypes.
Other Hereditary Neuropathies
Focal hereditary neuropathy variants include hereditary neuropathy with predisposition to pressure palsy (HNPP) and hereditary neuralgic amyotrophy. HNPP is an autosomal dominant neuropathy associated with liability to the development of focal neuropathies at common sites of compression. It is related to a deletion in the PMP22 gene on 17p11.2, the same gene that is duplicated in CMT1A. In a large cohort of patients with CMT, HNPP was responsible for 10% of cases. It can usually be distinguished from other forms of CMT by focal clinical signs and symptoms and electrophysiological features of multifocal demyelination at common sites of entrapment.
Hereditary neuralgic amyotrophy, also called hereditary brachial plexus neuropathy, is an autosomal dominant disorder associated with bouts of severe pain and motor greater than sensory deficits in the distribution of individual nerves of the upper limb and occasional cranial nerves. About 20% of cases are related to a mutation in the SEPT9 gene. Patients present with clinical features that overlap significantly with that of idiopathic neuralgic amyotrophy and distinguishing between idiopathic, and hereditary cases is often difficult. In addition to the previously mentioned neuropathies, there are numerous additional rare hereditary neuropathy variants that are beyond the scope of this article.
Hereditary myopathies
Dystrophinopathies
The dystrophinopathies include the spectrum of X-linked recessive disorders related to a mutation in the DMD gene located on chromosome Xp21.2-p21.1. Two allelic variants are commonly seen. DMD, the most common muscular dystrophy, affecting 1 in 3500 boys, occurs in individuals with severe deficiency of dystrophin. Onset occurs between 3 to 5 years of age with progressive, symmetric, proximal predominant weakness. Common clinical features include dilated cardiomyopathy, scoliosis, and mild mental retardation. Treatment with corticosteroids modifies disease progression. Becker muscular dystrophy (BMD) is a dystrophinopathy with a more variable clinical phenotype occurring 1 in 20,000 births. The onset is usually after 7 years of age, but the severity and age of onset is related to muscle dystrophin levels. Failure to walk may be variable, occurring from 16 to 80 years of age. Potential associated features include cardiomyopathy, mental retardation, muscle hypertrophy, and myalgia. Other phenotypes of dystrophinopathy include selective cardiomyopathy and manifesting female carriers. In selective cardiomyopathy (also termed X-linked dilated cardiomyopathy), patients present with dilated cardiomyopathy with minimal weakness. Female carriers may present with proximal weakness, cramps, and myalgia.
Genetic testing is the preferred method for diagnosis in the dystrophinopathies and it should be considered particularly in young boys presenting with proximal weakness before other invasive testing methods. Creatine kinase is typically markedly elevated and is a reasonable screening tool in individuals suspected to have dystrophinopathy. Demonstrating the presence of a pathogenic variant in the DMD gene provides molecular confirmation of the diagnosis. The lack of the identification of a mutation in the DMD gene lessens the chance of a dystrophinopathy, but sensitivity depends on the screening procedures used. DMD deletion/duplication testing will identify approximately two-thirds of affected males, but clinicians should be certain that the laboratory they select uses a method that interrogates all 79 exons in the gene, such as multiplex ligation-dependent probe amplification or comparative genomic hybridization. In cases in which a deletion or duplication is not identified and a consistent clinical phenotype is present, screening for point mutations or muscle biopsy with dystrophin quantification should be considered.
The major determinant as to whether mutations are associated with DMD or BMD is whether the mutations result in a preserved mRNA reading frame that allows translation of a protein with both N-terminal and C-terminal functionality. This rule, termed the reading frame rule, holds true in around 90% of cases of DMD and around 60% of BMD cases. These percentages demonstrate that the phenotype cannot be predicted by genomic analysis alone and reminds the clinician that prognosis should not be based on the genetic report in isolation.
Myotonic Dystrophies
Myotonic dystrophy is a highly variable, autosomal dominant disorder of skeletal muscle associated with multisystem involvement. It is the most common hereditary neuromuscular disease in adults. Two forms of myotonic dystrophy have been described: myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2) ( Table 2 ). Both disorders are characterized by clinical and electrical myotonia and progressive weakness. DM1 is characterized by myotonia and weakness that is most prominent in the distal limbs, bulbar, and neck flexors. DM1 is related to an expanded and unstable CTG trinucleotide repeat localized to the 3′ untranslated region of the dystrophia myotonica protein kinase ( DMPK ) gene located on chromosome 19q13.3. Associated systemic features include cataracts, excessive daytime sleepiness, insulin resistance, and impaired cognitive function. Because of repeat instability, DM1 is marked by repeat expansion in subsequent generations, with anticipation in disease onset and severity. The longest repeats are associated with congenital myotonic dystrophy, the most severe form of DM1, which is present at birth. Congenital DM1 is associated with hypotonia, weakness, and difficulties with breathing, sucking, and swallowing at birth. Motor function improves significantly in early life, but motor and self-care skills are often delayed. In children with congenital DM1, learning disabilities are common. Mutational analysis is available as a polymerase chain reaction–based test for the expanded DMPK allele, but clinicians should be aware that in some congenital cases the repeat size is too large for efficient amplification of the expanded allele, resulting in false-negative tests that require the use of a different testing modality to resolve.
| Myotonic Dystrophy Type 1 | Myotonic Dystrophy Type 2 | |
|---|---|---|
| Gene mutation | DMPK | ZNF9 |
| Weakness | Distal predominant | Proximal predominant |
| Myotonia | Usually prominent | Present but less severe |
| Pain | Unusual | Frequent |
DM2, also known as proximal myotonic myopathy, is related to a mutation in the CCTG repeat sequence in intron 1 of the ZNF9 gene. DM2 is associated with myotonia, proximal predominant weakness, and other systemic symptoms of cardiac dysfunction, insulin insensitivity, cataracts, and testicular failure. Myotonia is not usually severe, but pain is often prominent. Unlike DM1, a congenital form of DM2 has not been reported.
Limb-Girdle Muscular Dystrophies
Limb girdle muscular dystrophy (LGMD) is a descriptive term describing a large heterogeneous group of muscle disorders associated with proximal limb weakness. The clinical presentation of several types of LGMD resembles that of the dystrophinopathies but affects male and females equally. Multiple forms of LGMD with autosomal dominant inheritance classified as type 1 (LGMD 1) and recessive inheritance classified as type 2 (LGMD 2) have been identified. In about 75% of cases, genetic testing can be used to identify the specific form.
The general evaluation of possible LGMD include laboratory, electrodiagnostic, muscle biopsy, cardiac, and molecular testing. The history may provide clues to a particular form based on features of the age of onset, inheritance pattern, and other associated features. Examination, by definition, usually demonstrates proximal limb muscle weakness, but atypical features of distal limb weakness, focal weakness (ie, scapular winging), and contractures may be present and should be recognized. Creatine kinase levels may vary considerably and may be helpful in discerning between subtypes. Electrodiagnostic testing is helpful to determine the presence of a myopathic process and to exclude other conditions with proximal predominant weakness, such as spinal muscular atrophy (SMA) or congenital myasthenic syndrome, but electromyographic (EMG) findings are often nonspecific, with significant overlap between different forms of LGMD. Table 3 outlines some clinical and laboratory features that may be associated with each autosomal dominant and recessive form of LGMD.
Related posts:
 Neuromuscular Disease Management and Rehabilitation, Part I: Diagnostic and Therapy Issues
Electrodiagnosis in Neuromuscular Disease
Exercise in Neuromuscular Diseases
Physical Therapy Evaluation and Management in Neuromuscular Diseases
Robotics, Assistive Technology, and Occupational Therapy Management to Improve Upper Limb Function in Pediatric Neuromuscular Diseases
Clinical Approach to the Diagnostic Evaluation of Hereditary and Acquired Neuromuscular Diseases
Neuromuscular Disease Management and Rehabilitation, Part I: Diagnostic and Therapy Issues
Electrodiagnosis in Neuromuscular Disease
Exercise in Neuromuscular Diseases
Physical Therapy Evaluation and Management in Neuromuscular Diseases
Robotics, Assistive Technology, and Occupational Therapy Management to Improve Upper Limb Function in Pediatric Neuromuscular Diseases
Clinical Approach to the Diagnostic Evaluation of Hereditary and Acquired Neuromuscular Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


