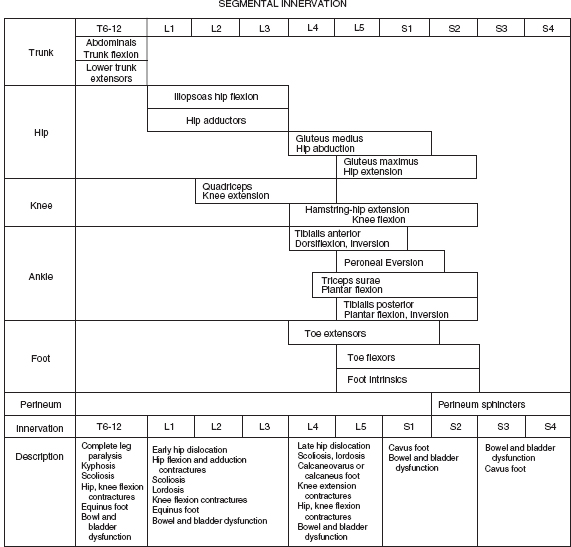
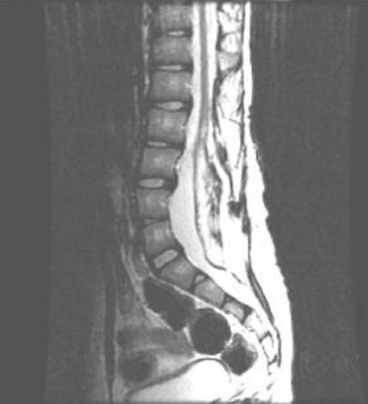
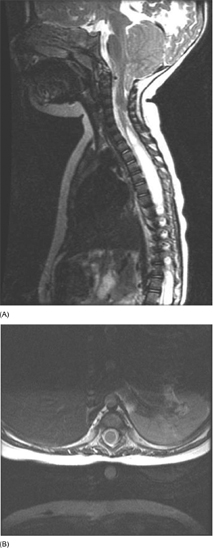
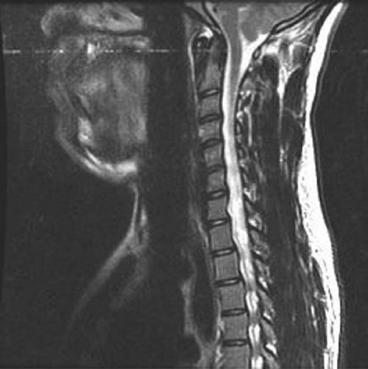
15 SPINA BIFIDA Elaine L. Pico, Pamela E. Wilson, Kathryn Smith, Jeffrey Thomson, Jeffrey Young, Daria Ettinger, Gerald H. Clayton, and Kerstin Sobus Spina bifida is the second most common disability in children. The National Spina Bifida Association (SBA) documents more than 70,000 individuals in the United States living with spina bifida. This is a small fraction of all those affected worldwide. Spina bifida is a complex disorder that has physical, psychological, and social implications. Medical professionals treating these individuals should have a thorough understanding of the spectrum of the disability. EPIDEMIOLOGY According to estimates by the Centers for Disease Control and Prevention (CDC), spina bifida and anencephaly, the two most common neural tube defects (NTDs), affect approximately 3,000 pregnancies yearly in the United States. NTDs vary in prevalence, depending on race and ethnicity, with women of African American and Asian descent having the lowest, while women of Hispanic ethnicity having the highest. It is notable that affected pregnancies in both Hispanic and non-Hispanic Whites have declined significantly since the mandatory fortification of grain products in the United States with folate (see the discussion in the section “Genetic Influences”). Folic acid fortification can substantially decrease a woman’s risk of an NTD-affected birth. Although the impact of folic acid on NTDs does not appear to be influenced by race or ethnicity, the disparity between Hispanics and other races and ethnicities remains, and the causes are unknown. It is hypothesized that differences in eating habits, supplement-taking practices, and in general an awareness of how nutrition affects pregnancy outcomes have a major impact on the developing fetus. Studies have demonstrated also that other risk factors such as maternal obesity, nutrient intake, and supplement use affect racial/ethnic groups differently. Genetic factors can have a major direct impact on NTD incidence and serve to alter susceptibility to numerous environmental influences. It is this interaction that requires continued study in order to discern how racial and ethnic factors change over time and affect NTDs. A study of spinal defect repairs from 1988 to 2010 showed a decrease in surgical correction; however, more operations were performed per 10,000 births for Hispanic patients as compared to Caucasian or African American members of this cohort (1). It has been suggested that folate fortification of corn masa flour may improve the known higher rate of myelomeningocele in the Hispanic population due to the prevalence of this flour’s use by this population (2). The current American Academy of Pediatrics (AAP) guidelines for folic acid supplements are: • All women of childbearing age: 400 μg or 0.4 mg/day. • Women who have spina bifida, or a female with a previous NTD pregnancy: 4,000 μg or 4 mg/day 1 to 3 months prior to conception and through the first trimester. This reduces the risk by 70% as per the SBA 2014 recommendations (3). • High-risk pregnancies (such as a mother who is taking valproic acid or has maternal diabetes): 4 mg/day (4). Understanding the multifactorial etiology of this disorder is complex, and even though much is known about the genetic errors associated with folate metabolism, other underlying genetic risks are still to be elucidated. Although incidence and recurrence risk vary around the world, most children are born to families without a prior affected child (0.1%–0.2%). The risk for recurrence in a family with one child with NTD is 2% to 5% and increases to 10% to 15% if two siblings are affected. If one parent has spina bifida, the risk of having a child with a similar disorder is 4% (5). It is important for the clinician to understand the embryogenesis of NTDs. The two forms of defects, open and closed, can occur at any point along the neuraxis with varying prevalence. Closed forms (encephalocele, meningocele, iniencephaly, lipomeningocele, sacral agenesis, and occult spinal dysraphism) and open forms (anencephaly, and myelomeningocele) may result from differing yet overlapping factors (6). Spina bifida is a complex, heterogeneous disorder whose etiology in humans appears to be multifactorial. However, spina bifida, classically defined as myelomeningocele, is the consequence of neural tube closure failure during embryonic development. The following section discusses the normal central nervous system (CNS) embryogenesis and the pathologic differences associated with these neural tube defects (7–10). NORMAL DEVELOPMENT During the first 2 weeks, postfertilization embryonic development involves repeated cell division and organization, resulting in a blastocyst, an embryo with two layers: the epiblast and the hypoblast. At the end of this period, on days 13 to 16, a primitive streak forms, beginning caudally and progressing toward the rostrally located prochordal plate initiating the rostral-to-caudal orientation of the embryo. The development of the primitive streak is followed by invagination of epiblast cells, which remodels the embryo (ie, gastrulation) into a three-layered structure comprising ectoderm, mesoderm, and endoderm, the precursors of all tissue types and body structures. As the primitive streak regresses, presumptive notochord cells migrate through Hensen’s node aligning themselves along the midline of the embryo between the endoderm and the ectoderm. In humans around day 16 the overlying ectoderm is induced by cell-to-cell contact with the notochord, thereby forming the neural plate. By about day 21, the plate bends as the groove deepens, and its walls and their adjacent cutaneous epithelium begin to oppose one another, beginning the formation of the brain and spinal cord. The eventual closure of the neural tube proceeds over a period of 4 to 6 days and typically involves primary closure of the cutaneous ectoderm. This is followed by the neuroectoderm, which then separates from the overlying cutaneous ectoderm, resulting in a closed tube. Closure begins at a point just caudal to the developing rhombencephalon and proceeds via several waves rostrally and caudally rather than in the continuous “zipper-like” fashion previously envisioned. There is, however, an alternative view proposed by Van Allen and colleagues that describes several closure initiation sites over the same period of time (11,12). Regardless, closure of the primary neural tube is typically complete around embryo developmental day 27. This process, primary neurulation, completes the presumptive spinal cord down to the lower lumbar and/or upper sacral levels. A secondary wave of neurulation begins around day 25 from a collection of remaining primitive streak cells and mesoderm located along the midline axis from the caudal end of the primary neural tube to the cloaca. Cavities form that coalesce into a tube, which eventually becomes continuous with the primary neural tube. This process completes the formation of the sacral levels of the spinal cord and terminal filum, and is species-specific. Understanding the process of primary and secondary neurulation is of paramount importance in comprehending the pathogenesis of spina bifida. The failure of primary neurulation is known to be associated with open spinal dysraphisms such as myelomeningocele, craniorachischisis, or anencephaly. Disruption of secondary neurulation, the mechanism for the development of the caudal spinal cord, is associated with closed spinal defects (eg, spina bifida occulta) and spinal cord tethering (13,14). The process of neurulation is completed by the end of the first month of embryonic development. Expansion of cranial brain structures via the development of a primitive ventricular system is thought to be accomplished by temporary occlusion of the caudal (spinal) neural tube on days 23 to 27, which creates a rostral-enclosed fluid-filled space. This provides pressure to expand the cranial lumen and is the impetus for brain enlargement. Theory suggests that, in part, failure of this expansion pressure is a cause for Chiari malformation (15) seen in spina bifida. PATHOLOGY Spina bifida is typically considered to be the result of a primary failure of neurulation. Failure of neurulation and, thereby, loss of neural tube closure, prevents the mesoderm, adjacent to the notochord, from forming muscle and bone (ie, via somitic mesoderm). This normally forms around the tube to protect it. Therefore, the mechanisms involved in this process are suspected to be involved in the pathology of this disorder. Although this is the most popularly accepted theory, there are other proposed mechanisms. Several forms of myelomeningoceles are not failures of neurulation, but a failure of Henson’s node to lay down the notochord correctly—in other words, a failure in gastrulation (16,17) that causes significant errors in the induction of the neural tube. Further research is necessary to verify current proposed theories. The mechanisms by which the neural tube is formed and closed are varied. Morphogenic changes in cell populations such as wedging and planar cell polarity result in the creation and shaping of the neural plate into a tube-like structure early on (18,19,20–22). Several other mechanisms are proposed for the closure of the neural tube, such as an interaction between various glycoproteins and cell adhesion molecules, various signaling protein/receptor interactions, the interlinking of numerous cell filopodia, and formation of intercellular junctions. The current view suggests the process likely involves all of these and perhaps others not yet visualized. Failure of any of these processes could therefore negatively impact CNS development. GENETIC INFLUENCES Genetic mutations have a significant impact on CNS development, as demonstrated experimentally in rodents and documented clinically in humans. Alterations in the genes that affect metabolism, nucleotide synthesis, cell programming, and cell-to-cell signaling can all affect aspects of neural development. This ranges from the signaling aspects of the induction of neural tube formation initiated by the notochord to alterations in apoptosis (programmed cell death). Induction of the neural plate is controlled by a variety of genes. Sonic Hedgehog (SHH) is a vertebrate gene expressed by cells within the notochord that—in conjunction with the Patched (PTC) gene—produce proteins involved in the induction of the floor plate during embryogenesis (23–25). During embryogenesis neuronal subtypes such as motor neurons proliferate, and somite development begins. The expression of various signaling proteins on the surfaces of cells allows for the sequential transmission of signals regulating the cell’s fate. PTC is a gene that functions downstream of SHH. Its function is hypothesized to serve as negative feedback to SHH, thereby regulating the induction of numerous cell types in the developing neural tube. Failure of this system and its feedback loop and/or overexpression of one portion of the process may lead to neural tube development failure. Although not currently implicated by empirical data, research is in place to elucidate the impact of this system on human NTDs. Alterations in genes that impact the movement of epithelial and mesenchymal cells that create and shape the body axis have also been suggested to be associated with NTDs. Planar cell polarity pathway genes such as SCRIB and PRICKLE may contribute to the multifactorial risk for spina bifida in humans (18,19). Genes that regulate folate metabolism and methyltransferase reactions associated with methionine and homocysteine metabolism are both of major interest (26). Folate serves as a cofactor for enzymes that participate in nucleotide synthesis, and is important in the methylation processes. Evaluations of folate levels of mothers with NTDs shortly after birth have produced equivocal results, suggesting that either absolute folate deficiency is rare or that their diet and folate level may have been different at the beginning of the pregnancy. However, disturbances in the metabolic pathways that utilize folate may predispose to NTDs. This cannot be corrected with folate supplements. Metabolism of folate and homocysteine are interdependent, and the risks associated with alterations in their metabolism are thought to be connected. Elevated homocysteine levels in pregnant women are a known risk factor for NTDs. Mutations/polymorphisms in the enzyme 5,10-methylenetetrahydrofolate reductase (MTHFR) have been associated with diminished plasma folate levels, with commensurate elevated homocysteine levels. These alterations have been identified in patients with spina bifida as well as in their parents. In addition, using cultures of fibroblasts from NTD-affected patients, homozygosity for defects in the MTHFR gene have been shown to have a 7.2-fold increased risk for NTDs. The prevalence of these defects appears to vary with race. Homozygosity for the C677T MTHFR mutation is a known risk factor for upper-level spina bifida lesions in those of Hispanic descent. The MTHFD1 1958G>A polymorphism is also associated with NTDs in those of Irish descent. Although folate supplementation has proven to be very successful in attenuating the prevalence of NTDs in various populations worldwide there is a portion resistant to such supplementation. Animal models show folate resistant NTDs are sensitive to the intake of inositol. Limited human experience has also suggested folate resistant NTDs may be amenable to inositol supplementation (27,28). In early stages of nervous system development, more cells are produced than needed and that the processes of apoptosis and autophagy are coordinated during development to yield a well-defined and functioning CNS. Apoptosis, the most studied of these processes, is modulated by various members of the Bc12 gene family, the caspase family of cysteine proteases, and other genes that produce necessary intermediator proteins. The variation of genetic expression controls the development of cells within the CNS. Several mouse models have shown altering the expression of these genes (ie, knockout experiments) results in NTDs similar to that identified in humans. Such evidence strongly suggests their involvement in human neural tube pathology. Autophagy, an autodegenerative cell process, has a significant impact on the recycling of cellular components in the cytoplasm as a result of cellular organelle damage. This process can also be affected by nutritional stresses. A variety of genes that affect this process have been investigated using mouse models. Loss of Beclin 1 and Ambra 1 expression has been noted to result in overgrowth of the developing CNS. Therefore, identifying the human equivalent of these and other similar genes could yield information as to the cause of various NTDs and provide information for new therapeutic targets. The external environment has a significant impact on embryonic development and the incidence of NTDs (29). Hyperthermia during early pregnancy, the first 28 days during neurulation, has been shown to increase the incidence of NTDs. Specifically, maternal febrile events as well as sauna and hot tub use have increased the risk of NTDs (30–34). Parental occupation has been demonstrated to have a definitive influence on the risk for NTDs. Increases in NTD risk have been noted for occupations involving exposure to solvents (eg, painters, industrial process workers, etc.). Health care, agricultural, and transportation workers have also been noted to have an increased NTD risk (35). The etiology of this occupational increase in NTD risk can only be hypothesized at this time. Nutritional influences have a broad impact on NTD risk and interact in many ways with environmental as well as genetic influences. As indicated earlier in this chapter, a primary example is folate metabolism. Folate is a cofactor for the enzymatic process involved in purine and pyrimidine synthesis and facilitates the transfer of methyl groups during the metabolism of methionine and homocysteine. Taken together, alterations in these folate-sensitive processes can have an impact on cellular proliferation. Lowered intake of foods containing folate in the diet is associated with an increase in NTD risk. Disorders of absorption of folate in the intestine can alter folate levels and potentially increase NTD risk. However, studies of folate receptor carrier densities in the intestines of women with NTD offspring or their progeny do not have abnormally low receptor levels (36). Studies have shown that supplementation can decrease prevalence. Mandatory supplementation of folate in grain products in the United States has caused a steadily declining NTD incidence since initiated in 1996. Since the introduction of mandatory folate supplementation, the number of NTD affected pregnancies has declined from approximately 4,000 to 3,000 per year. Studies have shown the NTD recurrence risk can be decreased by approximately 50% by taking the recommended folate supplementation. The risk for NTDs varies for couples, depending on whether there is a prior history of such defects. U.S. couples with a prior history of NTD births have an increased risk for recurrence (2%–5%) (5). Because of this, the U.S. Public Health Service and the CDC have two separate recommendations for supplementation based on prior NTD histories. Elevated supplementation is appropriate for couples with a prior NTD birth. Limited studies have also identified zinc deficiency as a nutritional entity that can also elevate NTD risk. It was discovered that women with the genetic disorder of zinc metabolism acrodermatitis enteropathica are at high risk for NTDs and that supplementation can lower those risks. Maternal obesity and concomitant diabetes are associated with increased NTD risk. Specifically, women with a prepregnancy body mass index (BMI) suggestive of obesity (>29 kg/m) are more inclined to have offspring with NTDs. There appears to be a direct relationship between maternal BMI and spina bifida (37). This holds true for women with diabetes, although the etiology of this association may be linked to alterations in glucose metabolism during organogenesis. It is notable that experimentally manipulated glycosylation in rodents results in birth defects not unlike those seen in children born to mothers with diabetes. Risks for NTD-affected births have been estimated at 2% in the United States and as high as 7% in England. These risks include spina bifida as well as other significant NTDs such as anencephaly. The NTD recurrence risk for mothers with diabetes in the U.S. is around 4%, which is similar to that found in mothers without diabetes. Teratogenic influences from the environment, such as the consumption of prescribed drugs, have been associated with NTDs, particularly myelomeningocele (38). Valproic acid taken for seizures during pregnancy has been shown to increase the incidence of NTDs. Valproic acid appears to work by disrupting folate metabolism, thereby inhibiting neural tube closure. An alteration in folate-dependent methylation of regulatory proteins is theorized to be the cause. Regardless, administration of folate during pregnancy counteracts valproic acid associated NTDs. Of note, the 2014 edition of the British HIV association guidelines indicate nonnucleoside reverse transcriptase inhibitors such as efavirenz should be acceptable for HIV infected women wishing to conceive (39). Earlier limited studies suggested against the use of antiretroviral agents such as efavirenz due to the apparent risk of NTDs; however, a meta-analysis by Ford and colleagues did not find an increased risk of overall birth defects associated with first trimester use (40). The authors caution due to small sample size and relative low NTD incidence, conclusions should receive further prospective evaluation. World Health Organization (WHO) and U.S. guidelines advise against first trimester use but can be considered later in pregnancy (41). Drug-induced interference with DNA synthesis during embryonic development would likely have an impact on gastrulation and neurulation. Other drugs are also associated with NTDs, such as isotretinoin (Accutane), used for acne treatment; and etretinate (Tegison), a psoriasis treatment; and anticancer agents such as methotrexate. Consumption of alcohol and coffee and cigarette use have been suggested to increase the risk of spina bifida. However, a recent study has reported confounding data (42). Fetal alcohol syndrome has an association with increased risk for abnormal CNS development, including NTDs. Ethanol-induced impairment of polyamine homeostasis has been suggested as a potential cause of NTDs and intrauterine growth restriction in fetal alcohol syndrome (43). PRENATAL SCREENING Prenatal screening is recommended for pregnant women to detect not only NTDs but also to screen for Down syndrome and other disorders. Prenatal diagnosis of spina bifida is important for families. It allows for decisions regarding continuation of the pregnancy, prenatal interventions, choice of appropriate birth facility, and preparation of parents and other family members for the birth to be made. Many parents choose to continue the pregnancy, although in a review of 17 studies Johnson and colleagues found that the overall frequency of termination of pregnancy ranged from 31% to 97%, and was more common for diagnosis confirmed before 24 weeks’ gestation, with defects of greater severity, and in Europe versus the U.S. (44). Prenatal diagnosis is generally carried out by one of several methods (45–48). A quad screen blood test is done in the second trimester. The test includes alpha fetoprotein (AFP), human chorionic gonadotropin (HCG), estriol, and inhibin A. Elevated levels of AFP suggest an NTD is present, and the need for further testing is indicated. The serum AFP test is not specific for spina bifida, however, and requires the gestational age to be correct. A study of open NTDs found maternal serum biochemical markers in the first trimester did not distinguish an open spina bifida. Further testing is required, using high-resolution ultrasounds (US) and amniocentesis (49). The fetal US helps identify characteristics both of the brain and the spine, such as an abnormal shape of the head, enlarged ventricles, or abnormal vertebrae. US can detect a splaying of the pedicles and the classic “lemon and banana signs.” The lemon sign relates to the shape of the head, and the banana sign is related to herniation of the cerebellar vermis through the banana-shaped foramen magnum. Amniocentesis can also be performed, typically at 15 to 20 weeks of gestation, to identify abnormally high levels of AFP. Fetal MRI, after diagnosis, is made to determine the extent of the defect. Prenatal diagnosis allows for the ability to plan ahead. Fetal surgery has been available for families on a research basis through the Management of Meningomyelocele Study (MOMS) program comparing surgery before (in utero) and the standard surgery after birth. A discussion of in utero closure is beyond the scope of this chapter, but the procedure is carried out only in specialized settings. The MOMS study is ongoing, but not recruiting participants at the time this goes to print. Please refer to the website: http://clinicaltrials.gov/show/NCT00060606. Differences of opinion exist as to the preferred delivery method, vaginal or cesarean section (previously preferred method of delivery). The delivery should occur at a high-risk center with access to a neonatal intensive care unit and a full range of specialists, in neurosurgery and urology. CLINICAL TYPES OF NEURAL TUBE DEFECTS SPINA BIFIDA OCCULTA • Bony defect with no herniation of meninges or nervous elements • Incidental finding in 5% to 36% of adults; a small percentage can develop clinical findings (50,51) • Can be associated with pigmented nevus, angioma, hairy patch, dimple, and dermoid sinus • Usually found in the lumbosacral/sacral segments • Can have associated tethered cord with development • May have bowel and bladder involvement • No hydrocephalus or Chiari malformation SPINA BIFIDA CYSTICA • Bony defect with herniation of spinal canal elements • Meningocele herniation of the meninges, but does not contain neural tissue • Meningomyelocele herniation of meninges and neural elements CAUDAL REGRESSION SYNDROME • Absence of the sacrum and portions of the lumbar spine • Associated with maternal diabetes • Associated findings include syringomyelia, anorectal stenosis, renal abnormalities, external genital abnormalities, and cardiac problems • Motor and sensory abnormalities The spinal cord defect associated with spina bifida is often associated with other malformations. This results in a multisystemic process that leads to a variety of health problems and potentially life-threatening complications. Motor and sensory deficits vary according to the level and extent of spinal cord involvement (52–54). In the care of individuals with spina bifida, two levels are often described: the anatomic level of the lesion and the neurologic level of functional involvement. In terms of the level, it is the neurologic or functional level that gives health care providers prognostic information with respect to long-term expectations and functional outcomes. Spinal cord involvement may result in asymmetric motor and sensory deficits. Sensory deficits usually follow a dermatomal pattern and may not affect all sensory modalities equally (52,53). Neurogenic bladder and bowel dysfunction may be present in all patients because of the distal level of innervation of the bladder and bowel. This is true even if there is no apparent motor deficit in the legs. In the following discussion, clinical signs of muscle weakness are described. These functional neurologic levels may not directly reflect the anatomic level of the malformation. Musculoskeletal deformities related to muscle imbalance may present serious clinical concerns. Deformities may be static deformities present at birth or may develop over time. Figure 15.1 summarizes segmental innervation, preserved muscle function, and musculoskeletal complications typical of various levels of spinal cord malformation. Providers must keep in mind that the overall functional outcome for the individual is related in part to neurologic level, in addition to other associated CNS, psychosocial, and medical issues. THORACIC LESIONS Thoracic-level malformations spare the upper extremities, with the exception of decreased ability to abduct the fifth digit (thoracic level 1 = T1). There is usually partial innervation of the abdominal and intercostal musculature, which may result in respiratory dysfunction or insufficiency. Kyphosis and kyphoscoliosis may result from trunk weakness and be more prominent in individuals with vertebral anomalies (55). The legs may be flaccid or show signs of spasticity when some portion of the distal spinal cord is preserved (56). The lack of volitional movements combined with the effect of gravity leads to lower extremity deformities. The usual lower extremity posture in the supine position is partial hip external rotation, abduction, and ankle plantar flexion from gravity. Knee flexion contractures and equinus foot deformities develop from sitting. Hip flexion contractures with compensatory lumbar lordosis increase any pre-existing kyphosis or kyphoscoliosis (56). L1–L3 SEGMENT Hip flexors and hip adductors are innervated at the L1 to L2 levels. With L2 sparing, knee extensors have partial innervation but are not at full strength. The distal lower extremity muscle strength is absent. The distribution of muscle imbalance—hip flexion and hip adduction with absent hip extension and hip abduction—leads to the development of contractures and early paralytic hip dislocation. Pelvic obliquity seen in asymmetric hip pathology enhances scoliosis. Gravity-related foot equinus deformity may develop (56). Ambulation during young childhood is typical with the use of bracing and assistive devices. Long-term ambulation through adulthood is less likely as priorities change and there are further increases in the already high-energy demands of walking (54). The extent of bracing necessary to achieve ambulation is usually related to the amount of active knee extension. L4–L5 SEGMENTS Innervation of the hip flexors, hip adductors, and knee extensors is usually complete; however, hip abductors and hip extensors remain weakened. Coxa valga and acetabular dysplasia are still a concern. Typically, hip dislocation occurs later at the L4 to L5 segmental levels. Newborns with a well-defined lesion sparing L4 lie in a typical position of hip flexion, hip adduction, and knee extension. When the L5 segment is spared, the gluteus medius, gluteus maximus, and hamstrings have partial strength and knee extensor contracture is less likely. Because the tibialis anterior is unopposed by its plantarflexion and everter antagonists, a calcaneovarus foot deformity develops. If the peroneus muscles are spared, the varus is eliminated. Although the plantar flexors are partially innervated, they are not strong enough to counter the strong force of the ankle dorsiflexors (56). SACRAL SEGMENTS Active plantarflexion is stronger and some toe movements are present. Intrinsic foot muscles remain weak and may result in a cavus foot deformity with clawing of the toes (56). FIGURE 15.1 Musculoskeletal, sensory, and sphincter dysfunction by segmental level. SENSORY DEFICIT Partial or complete absence of sensation predisposes individuals with spina bifida to skin injuries because of decreased ability to perceive pressure, pain, trauma, or heat (52–54,57). Skin breakdown tends to occur over areas of prominence and weight-bearing. The lower back, intergluteal, perineum, feet, heels, and toes are the sites of predilection, but any area with sensory loss may be affected. Scoliotic and kyphotic prominences are areas prone to breakdown (55). Pressure ulcers often heal slowly, tend to get infected, and often recur. A refractory pressure ulcer may be a symptom of a tethered cord. Long-standing ulceration with deep tissue necrosis may spread to bone and lead to acute or chronic osteomyelitis. Weight-bearing is often on the anesthetic heel, and a deep ulcer can lead to osteomyelitis of the os calcis (56). Other complications of denervation include vasomotor instability, neuropathic Charcot joints, and osteoporosis in individuals with extensive lower extremity weakness (54,55,57,58). Sensation may be decreased with regard to sexual function thus causing different issues for males and females. Please see the section on Long Term Aging With a Neural Tube Defect where this is discussed in more detail. ASSOCIATED CENTRAL NERVOUS SYSTEM MALFORMATIONS Extensive neuropathologic studies have demonstrated that NTDs are associated with a high incidence of gross and microscopic malformations of the forebrain and hindbrain (59). Additional anomalies in the spinal cord may complicate the original local dysraphic defect (52,53,57,60,61). Table 15.1 lists associated anomalies and malformations by location. SPINAL CORD The spinal cord defect usually results in a lower motor neuron process. Spasticity is present in most individuals with spina bifida across their lifetime (62). The presence or gradual development of spasticity above the level of the spinal cord lesion may be related to tethering of the spinal cord, Chiari type II malformation exerting pressure on the cervical spinal cord, decompensating hydrocephalus, ventriculitis, syringohydromyelia, or coexistent encephalopathy sustained at birth (63–66). TABLE 15.1 ASSOCIATED CENTRAL NERVOUS SYSTEM MALFORMATIONS Spinal cord Tethering Distal focal abnormalities Thick, short filum terminale Supernumerary fibrous bands Lumbosacral tumors (lipoma, fibrolipoma, fibroma dermoid, epidermoid cyst, teratoma) Bony vertebral ridge Diastematomyelia, diplomyelia, split cord Brainstem Arnold type II malformation Kinking, inferior displacement of medulla Herniation into cervical spinal canal Abnormalities of nuclear structures Dysgenesis, hypoplasia, aplasia, defective myelination Hemorrhage, ischemic necrosis Syringobulbia Cerebellum Arnold-Chiari type II malformation Elongated vermis, inferior displacement Herniation into cervical spinal canal Abnormal nuclear structures Dysplasia, heterotopia, heterotaxia Ventricular system Hydrocephalus Aqueductal stenosis, forking, atresias Forebrain Polymicrogyria Abnormal nuclear structures Heterotopia (subependymal nodules) Heterotaxia Prominent massa intermedia Thalamic fusion Agenesis of olfactory bulbs and tracts Attenuation/dysgenesis of corpus callosum Tethered cord refers to an abnormal attachment of the spinal cord at its distal end (57). Under normal circumstances, the conus medullaris ascends from its distal position to the L1 to L2 vertebral level during the first year of life (67). Focal abnormalities—including thickened and shortened filum terminale, supernumerary fibrous bands, persistent membrane reunions, dural sinus, diastematomyelia, entrapment by lumbosacral tumors, and adhesions in the scar tissue of the repaired myelomeningocele—interfere with this process (57). Almost all children born with spina bifida have a low-lying cord on MRI, and approximately one-third develop neurologic, urologic, or orthopedic complications or symptoms (68) (Figure 15.2). Tethering of the spinal cord is the second most common cause of neurologic decline in children with myelomeningocele (68). The most common clinical signs or symptoms of a tethered cord include spasticity in the lower extremities, decline in lower extremity strength, and worsening scoliosis. Other signs and symptoms that strongly suggest tethering of the spinal cord include back pain, changes in urologic function, changes in gait, and development of lower extremity contractures. In patients who are suspected of having a symptomatic tethered cord, the function of their shunt must be evaluated prior to proceeding forward with surgical management (69), as shunt malfunction can present similarly. FIGURE 15.2 T2-weighted magnetic resonance image of tethered cord. There is tethering of the spinal cord with conus seen down to the L5 vertebral level, heterogeneous signal intensity characteristics, and areas of fibrofatty tissue. Source: Molnar GE. Spina bifida: clinical correlations of associated central nervous system malformations. Phys Med Rehabil State Art Rev. 1991;5:289, with permission. Diastematomyelia is a postneurulation defect that results in a sagittal cleavage of the spinal cord, most commonly affecting the lumbar and thoracolumbar levels of the spinal cord. It is more common in females (73,74). Diastematomyelia may have both neurologic and orthopedic presentations. Orthopedic symptoms include scoliosis, Sprengel’s deformity (especially when associated with Klippel–Feil sequence), hip subluxation, and lower extremity limb-length discrepancies (74,75). Neurologic symptoms include gait abnormalities, asymmetric motor and sensory deficits of the lower extremities, and neurogenic bladder and bowel (76). Symptoms of diastematomyelia may present in childhood or, less commonly, in adulthood (77). It is not uncommon for individuals to develop syringomyelia—a tubular cavitation in the spinal cord parenchyma extending more than two spinal segments (78). Syringomyelia is present in up to 40% of individuals with myelomeningocele (79). The syrinx may be located anywhere along the spinal cord, medulla, or pons, but is most common in the cervical spinal region (52,54,80). MRI is used to detect syringomyelia (81) (Figure 15.3). Often, a syrinx is of little clinical significance; however, if a patient develops decreasing function above the level of his or her lesion, syringomyelia must be considered in the differential diagnosis. Although shunt malfunction and spinal cord tethering are more common complications, symptomatic hydromyelia may explain a slower-than-expected progression through gross motor and fine motor developmental milestones or a decrease in strength or change in function. Early progression of scoliosis above the initial neurologic level may be the earliest sign of a syrinx. A shunt malfunction may contribute to a symptomatic syrinx, and shunt function should be evaluated. Placement of a syringopleural shunt may be necessary to decompress the syrinx. FIGURE 15.3 T2-weighted magnetic resonance image showing sagittal (A) and axial (B) views. There is a large syrinx present, beginning at the mid portion of C6 and extending to L4. The most common hindbrain abnormality in NTDs is Chiari type II malformation, seen in 80% to 90% of individuals with myelomeningocele (52,53,57,82). This malformation results in caudal displacement or herniation of the medulla, lower pons, elongated fourth ventricle, and cerebellar vermis into the cervical spinal cord (Figure 15.4). This often interferes with cerebrospinal fluid (CSF) outflow and is, therefore, almost always associated with hydrocephalus. Caudal displacement of the medulla may occur and result in traction neuropathies of the lower cranial nerves. Signs of bulbar compromise arise from compression of the herniated hindbrain. A broad spectrum of clinical symptoms is seen in individuals with this malformation. However, only 20% will develop clinical signs of brainstem dysfunction, with most occurring in the neonatal period (83,84). Symptoms may be evident at birth or present within the first 2 to 3 months. The most severe symptom is respiratory compromise, which may be both centrally and peripherally induced. Individuals may experience stridor, laryngeal nerve palsy with vocal cord paralysis, upper airway obstruction, periodic breathing, central or obstructive sleep apnea, or aspiration. Dysphagia and extraocular motion abnormalities, which are related to cranial neuropathies, may also be seen. Dysphagia may be severe enough that gastrostomy tube placement is required. Airway compromise may necessitate tracheostomy. FIGURE 15.4 T2-weighted magnetic resonance image of the cervical spine with Chiari II malformation. The posterior fossa is crowded. There is cerebellar tonsillar herniation, with the cerebellar tonsils lying 9 mm below the foramen magnum. In the presence of brainstem compromise, hemiparesis or tetraparesis may occur (this is more common in older children and adults than in infants). Impairment of fine motor hand function is well documented and is seen in more than half of individuals with thoracic-level lesions and approximately one-fourth of individuals with lumbosacral lesions. Control of ocular motility (saccadic eye movements, visual fixation, and pursuit) is related to cerebellar function. Fewer than one-third of individuals with spina bifida have completely normal visual function (85,86). Despite successful initial treatment with surgical decompression, problems may recur. Typically, vocal cord paresis in the first 2 months of life is a sign of irreversible damage, and surgical decompression is unlikely to result in clinical improvement (87). VENTRICLES Hydrocephalus is a significant problem in the majority of patients with myelomeningocele. The pathogenesis of hydrocephalus is multifactorial and is related to aqueductal stenosis, occlusion of the foramen of Luschka and Magendie, hindbrain herniation, obliteration of the subarachnoid spaces at the level of the posterior fossa, compression of the sigmoid sinuses with consequent venous hypertension, and fibrosis of the subarachnoid spaces (88,89). The prevalence of hydrocephalus in individuals with myelomeningocele is reported to be as high as 95%, with shunt rates ranging from 77% in the 1980s to 58% in more recent years (90). In a 23-year period from 1988 to 2010, the national trend was 56.6 % required surgical repair perinatally at the same time as shunt placement (91). Hydrocephalus rates are closely associated with the level of the spinal dysraphism. In one cohort, 100% thoracic, 87% lumbar, and 67% sacral myelomeningocele patients required shunting (92). In all cases of symptomatic hydrocephalus, surgical management is recommended. Symptoms of hydrocephalus include those that are classic for increases in intracranial pressure. This varies based on the presence or absence of an open fontanelle. In an infant, signs of increased intracranial pressure include lethargy, decreased feeding, bulging fontanelle, increasing head circumference (greater than expected for age), poor developmental progress, and “sun downing.” In patients with a closed fontanelle, signs of increased intracranial pressure include headache, vomiting, drowsiness, changes in behavior, changes in personality, irritability, diplopia, and papilledema. With the sudden onset of increased intracranial pressure, Cushing’s triad may be seen. Cushing’s triad consists of progressively increasing systolic blood pressure, bradycardia, and irregular respirations. Presently, placement of a shunt is the standard of care for surgical management of hydrocephalus. Shunting has many complications, including both mechanical and infectious. Up to 95% of adult patients with myelomeningocele have required at least one shunt revision. The rate of shunt infection is between 5% and 8% per procedure (68,93–97). Endoscopic management of hydrocephalus is being increasingly presented as an alternative to shunting. Endoscopic third ventriculostomy (ETV) provides direct communication between the third ventricle and the subarachnoid space by way of interpeduncular and prepontine cisterns. The success rates for ETV, as the sole management for hydrocephalus in infants with myelomeningocele, range from 12% to 53% (98–102). More recently, ETV has been combined with choroid plexus cautery (CPC). This has resulted in an improved success rate of greater than 70% for treatment of hydrocephalus in infants. Failure of ETV combined with a CPC typically is within the first 3 months (103). ETV may also be an option in the setting of a shunt malfunction in the older child. In one study the majority of ETV failures were during the first 6 weeks postoperatively. However, failures were seen as late as 5 years postoperatively (102). Longevity of the ETV/CPC for treatment of hydrocephalus beyond 2 or 3 years has yet to be determined. It is not known if there is a difference in neurocognitive outcomes in patients treated with an ETV/CPC (shunt-independent) as compared with individuals who are shunt-dependent. Although not yet considered the “standard of care,” ETV in combination with CPC holds promise for surgical management of hydrocephalus without creating shunt dependency and the associated complications (103). FOREBRAIN Malformations of the forebrain are broad, and range from gross anatomic malformations to microscopic anomalies. Polymicrogyria are increased numbers of small-sized cerebral gyri with shallow disorganized sulci, and this is seen in up to 65% of individuals (104). Heterotopias are aberrant neural tissues in the form of subependymal nodules. They are present in approximately 40% of cases (59). Microscopic studies have demonstrated disordered cortical lamination, neuronal hypoplasias of the thalamus, and complete or partial agenesis of the olfactory bulbs and tracts. Dysgenesis or agenesis of the corpus callosum may be seen and may also be associated with a malformed cingulated gyrus and septum pellucidum (104). The contribution of these forebrain malformations to the development of cognitive and perceptual dysfunction remains unknown. OTHER MALFORMATIONS NTDs are also associated with an increased rate of malformations unrelated to the CNS. Vertebral anomalies are not uncommon and contribute to progressive kyphosis and scoliosis (56). Thoracic deformities may result from rib deformities, including absence, bifurcation, or reduction of the ribs. Malformations of the urinary system may be present and result in accelerated deterioration of renal function. NTDs have been associated with genetic abnormalities, including trisomy 18, trisomy 13, Turner’s syndrome, Waardenburg’ s syndrome, renal aplasia and thrombocytopenia syndrome, nail-patella syndrome, 13q deletion syndrome, and others (105). TREATMENT TEAM APPROACH A team approach is an important part of the care of the individual with congenital spinal dysfunction. The multidisciplinary team often includes neurosurgery, orthopedic surgery, urology, rehabilitation medicine, physical and occupational therapy, social work, nursing, nutrition, and neuropsychology. Coordination of all modes of treatment is important for a successful rehabilitation plan. Primary care for the usual childhood illnesses and health maintenance including immunizations should remain the responsibility of the pediatrician. After birth, parents and families of individuals with spina bifida need to be informed about their child’s diagnosis and its implications. A prenatal visit with the neurosurgeon and other medical specialists is recommended. Parents often ask questions regarding anticipated functional abilities and limitations, including self-care and ambulation. Cautious predictions based on the current functional level should be given. Medical providers should be factual in their discussion of the problems that the parents and child will face. Discussions and instructions about the child’s care and handling at home may require several sessions so that the family members are not overwhelmed by the amount and complexity of the information. Families should be informed of the many issues involved and the need for seeing several medical specialists. Frequent follow-up after discharge from the neonatal unit is often necessary and typically involves visits every 3 to 4 months for a couple of years and then every 6 months thereafter (56). NEUROSURGICAL TREATMENT Involvement in the care of the individual with spina bifida begins with a prenatal visit to the neurosurgeon. Studies regarding prenatal surgical closure of a NTD are under way. To date, intrauterine repair has not been shown to decrease the motor deficits associated with myelomeningocele, but in some series it has been demonstrated to decrease the degree of associated Chiari type II malformations and the need for shunting procedures for hydrocephalus in the first year of life (106–109). Neurosurgical repair of an open NTD, such as a cystic lesion, is usually performed on the first day of life. If hydrocephalus is present at birth, surgical management may be necessary. Ninety-five percent of children with spina bifida are likely to have hydrocephalus, and 75% to 85% require surgical management. The average revision rate is 30% to 50% (110), and after 2 years of age there is a 10% per year risk of failure (96). Most neurosurgeons believe that a child with hydrocephalus that required shunting will remain shunt-dependent (97,111). These statistics may change as ETV with CPC is performed with increasing frequency. Neurosurgical follow-up is required, even after the neonatal period, to monitor for symptomatic hydrocephalus, shunt malfunction, and other neurosurgical complications. Pediatric patients with myelomeningocele should be followed routinely, usually on an annual basis. NEUROGENIC BLADDER A neurogenic bladder is found in most individuals with spina bifida. Data from the CDC National Spina Bifida Registry presented at the American Association of Developmental Medicine 2014 annual conference, reported 94% occurrence of neurogenic bladder in individuals with myelomeningocele with an over 60% urinary incontinence prevalence in those above 5 years. Furthermore, most with myelomeningocele use a catheter to empty their bladders. From the newborn period, the successful treatment of a neurogenic bladder is accomplished by the team approach including the urologist, physiatrist, nursing, and family. PHYSIOLOGY The fundus is made up of three layers of crisscrossing smooth muscle, called the detrusor. These three smooth muscle layers extend down the proximal urethra and stop at the external sphincter, which comprises skeletal muscle. T10 to L1 supplies the sympathetic innervation for the bladder; this causes the detrusor to relax and the bladder neck and posterior urethra to contract, effectively allowing storage of urine. S2 to S4 provide the parasympathetic innervation to the bladder primarily at the fundus and the neurotransmitter is acetylcholine causing contraction to empty the bladder. The sympathetic innervation is active during bladder filling, and the parasympathetic innervation is active during urination. Somatic nerves via the pudendal nerve (from the sacral cord S2–S4) innervate the skeletal muscle component of the external urethral sphincter: contraction occurs in the resting state for continence, and relaxation should occur during urination (112). BLADDER CAPACITY The prediction of normal bladder capacity aids the diagnosis of abnormal voiding patterns. It is typically accepted that the bladder capacity of a baby during the first year equals the weight of the baby in kilograms times 7 to 10 mL. A study with 200 children (132 with normal voiding, 68 frequent and infrequent voiders) demonstrated that from approximately 1 to 12 years of age, the sum of age in years plus 2-equals the bladder capacity in ounces (30 mL = one ounce). Teenagers (if normal size) assume adult-size bladders, typically around 400 to 500 mL. Clinically infrequent voiding may cause an increase in bladder capacity over normal values. Clinically frequent voiding may cause a decrease in functional bladder capacity (113). Postvoid residual (PVR) is generally accepted as less than 20% of bladder capacity, taking into account the appropriate bladder capacity for age. DIAGNOSTICS Checklist for Diagnosing Neurogenic Bladder • Is the current management preserving the kidneys and renal function? • If present, does bacteriuria represent infection or colonization? • Are the bladder and kidney studies up to date? • Is urination true voiding or overflow incontinence? • Are bladder capacity and bladder compliance appropriate for age? • Is PVR appropriate? • Is the sphincter mechanism competent? DIAGNOSTIC TESTS Urinalysis The nitrate test indirectly detects urine bacteria with enzymes that reduce nitrate to nitrite in urine (eg, Klebsiella, Enterobacteriaceae, Escherichia coli, and Proteus). THE LEUKOCYTE ESTERASE TEST. While leukocytes in the urine can disintegrate and disappear rapidly, leukocyte esterase persists. Urine culture (UC): Culture of urine for suspected clinical infection. Serum creatinine: Indicator of renal function, but typically lags renal function by several days. FUNCTIONAL STUDIES • US—Ultrasound of the kidneys and bladder to determine any structural abnormalities, including hydronephrosis, trabeculation, stones, and PVR. If not on catheterization program, consider US to measure PVR. • VCUG—Voiding cystourethrogram to detect vesicoureteral reflux (VUR), evaluate the bladder contour, and evaluate the urethra. For those without neurogenic bladders, the first study is a contrast VCUG for boys and girls. Subsequent VCUG studies, for boys and especially girls, can be radionuclear cystograms, as the radiation is reduced. However, for those with neurogenic bladders the fluoroscopic VCUG with contrast demonstrates greater detail of the bladder neck on all subsequent studies. Many centers perform cystograms as part of the urodynamic study, called videourodynamics. If videourodynamics are used, then a separate VCUG is not necessary. • UDY—Urodynamics to determine detrusor leak point pressure, uninhibited bladder contractions now called detrusor over activity (114), detrusor sphincter dyssynergia, bladder capacity, PVR, and bladder compliance and sensation. The basic urodynamic formulas are: Pressure detrusor = pressure vesical (bladder) − Pressure abdominal (rectum). On urodynamic testing, detrusor leak point pressure (p vesical − p abdominal) greater than 40 cm H2O, with a bladder capacity less than 33% of expected, was associated with renal damage (115). Bladder compliance = change in bladder volume/change in pressure It is recommended that these tests (US, UDY, and VCUG) be performed in the neonatal period, as baseline studies. The VCUG can be excluded if videourodynamics are utilized. As growth of the infant is rapid in the first 12 months, US should be repeated every 3 to 6 months during infancy. Greater frequency is encouraged if initial studies are abnormal. A common protocol incorporates US twice yearly the second year and then yearly. Abnormalities on US will likely lag those found on UDY. UDY should be repeated during the first year if the initial study shows a high detrusor leak point pressure of greater than 40 cm H2O or detrusor sphincter dyssynergia. Many centers perform UDY yearly in all patients to aggressively detect potential bladder deterioration that could harm the kidneys, whereas others repeat only if renal changes are noted on US or continence is not obtained by school age (116). VCUG (or video UDY) are needed if VUR was noted or if there is a new onset of recurrent UTIs or hydronephrosis. UDY studies should be repeated with significant clinical changes in bowel or bladder incontinence, infections, or gait, all of which are potential signs of secondary tethered cord (117). Other Studies RENAL STONES. The excretory urogram (EXU) also known as intravenous pyelogram (IVP), once used to detect urinary tract (UT) stones, anatomic abnormalities, and obstruction, is rarely done anymore. Following diagnostic US for renal stones, the EXU has been replaced by CT without contrast for more precise imaging. ANATOMIC VARIANTS. A magnetic resonance urogram with gadolinium is now used to demonstrate rare anatomic variants, particularly to delineate the ureters if ectopia is suspected. Diethylene triamine acetic acid (DTPA) and mercaptoacetyltriglycine (MAG3) are nuclear medicine studies used to evaluate differential function and excretion of each kidney; however, the MAG3 offers better imaging than DTPA to evaluate UT drainage/obstruction. The DTPA and MAG3 radioactive tracers are filtered (DTPA) or excreted (MAG3) into the renal tubules. Dimercaptosuccinic acid (DMSA), a radiotracer that binds to the tubule, is not rapidly excreted, and it is the best test for imaging the functioning renal cortex to detect congenital hypoplasia or acquired scarring. This test should be done when there is abnormality on a renal US, a history of multiple urinary tract infections (UTIs), or pyelonephritis. Cystoscopy is recommended for persistent hematuria or bladder masses. There is controversy regarding the role of routine screening cystoscopy in patients with increased risk of malignancy due to either bladder augmentation surgery or long-term indwelling catheter use (118,119). Figure 15.5 depicts a normal urodynamic study and Figure 15.6 is a tracing of a urodynamic study reflecting spastic bladder detrusor and sphincter dyssynergia. RISK FACTORS FOR UPPER TRACT/KIDNEY DETERIORATION • Detrusor leak point pressures greater than 40 cm H2O determined by UDY • Vesicoureteral reflux determined by VCUG/videourodynamics • Detrusor sphincter dyssynergia determined by UDY FIGURE 15.5 Normal urodynamic study. FIGURE 15.6 Urodynamic study reflecting spastic bladder detrusor and sphincter dyssynergia. • Poor bladder compliance determined by UDY • Detrusor overactivity previously known as bladder hyperreflexia • Increased PVR greater than 20% of the total bladder capacity TREATMENT Goals • Preservation of renal function • Age—appropriate social continence • No significant UTIs • Normalized lifestyle TREATMENT OF STORAGE DYSFUNCTIONS Excessive storage pressures due to detrusor overactivity previously known as detrusor hyperreflexia, or decreased compliance, can be decreased with anticholinergic medications. Oxybutynin chloride is commonly used and is available as syrup, and in immediate-release and extended-release tablets [Note: The youngest child in the study was 6 years old (120)]. Oxybutynin has been used intravesically, but is an off-label use. There is a transdermal form of oxybutynin as well as other anticholinergic medications on the market, but these are not currently Food and Drug Administration (FDA) approved in children. Botulinum toxin A (off-label) can be injected into the detrusor muscle via a cystoscope for those whom anticholinergic medication is ineffective. Electrical stimulation of the bladder directly or of innervating nerves has shown some promise to improve storage characteristics but remains experimental. Stress incontinence due to bladder outlet weakness caused by poor innervation of the internal and/or external urinary sphincters is more difficult to treat medically. Internal urethral sphincter resistance may be improved by the following alpha-sympathetic stimulation medications: phenylephrine, pseudoephedrine, and imipramine. External urethral sphincter weakness may improve with neuromuscular reeducation, but surgical treatment is typically required. TREATMENT OF EMPTYING DYSFUNCTIONS The typical day-to-day management of the neurogenic bladder in a majority of individuals with spina bifida is clean intermittent catheterization (CIC) every 4 hours while awake to keep bladder volumes within normal limits for age (113). In 1972, Lapides was the first to publish that the sterile single-use catheter is unnecessary in the management of persons with neurogenic bladders because it does not reduce bacteriuria (121). This continues to be substantiated in the pediatric population (122). If this intervention is unsuccessful, various pharmacologic and urologic surgical procedures may be explored. The Credé maneuver should be used with extreme caution. Using the Valsalva or Credé maneuver to empty the neurogenic bladder that has detrusor sphincter dyssynergia will likely raise the intravesicular pressure to greater than 40 cm H2O, thus putting the kidneys at risk. Bethanechol is rarely used to treat weak expulsive force of the detrusor because emptying is typically incomplete and synergistic sphincter relaxation does not occur. Hyperactive internal sphincter mechanism may be treated with alpha-adrenergic blockers. Hyperactive external urethral sphincter may be treated with baclofen, neuromuscular reeducation of the pelvic floor, Botox injections (off label) (123), or surgery. The Mitrofanoff procedure, first introduced in the 1980s, creates a continent catheterizable channel, between the bladder and the umbilicus/or lower abdomen. The appendix is most commonly utilized to create the channel, but other structures, such as a refashioned small bowel or ureter can be used. A tunnel between the bladder mucosa and detrusor muscle should provide sufficient compression to prevent external leakage; however, leakage may be problematic in a small percentage (124) but most should be continent. A Mitrofanoff procedure may be useful for patients unable or unwilling to cath per urethra, especially in females who may have more difficulty catheterizing than males. The procedure is commonly performed in the setting of bladder outlet surgery that usually prevents leakage per urethra but makes urethral catheterization more difficult. A vesicostomy may be a temporizing measure for younger children to avoid deleterious high bladder pressures. The ileal conduit, an incontinent urostomy to an external bag was commonly utilized in children with spina bifida prior to the 1970s before both CIC and oxybutynin were introduced. Follow-up studies showed a disappointingly high rate of renal deterioration, calculosis, hydronephrosis, and the need for reversal of the ileal conduit procedure (56). Artificial urethral sphincters have been found to be helpful in some, but can cause infection, erosion, and mechanical problems. PRIMARY CARE TREATMENT OF CHILDREN MANAGED WITH CLEAN INTERMITTENT CATHETERIZATION Routine urinalysis (UA) and UC are not recommended during well-child check-ups if the child looks well and is asymptomatic. If bacteriuria is detected in the urine, it is important to determine whether it represents a clinical infection or colonization of the bladder. Only clinical UTIs with symptoms of fever, abdominal/back pain, painful urination/catheterization, new or worsening incontinence or urinary symptoms, or hematuria should be treated (122). Prophylaxis in the absence of VUR is not routinely recommended. ANTIBIOTIC PROPHYLAXIS AND BACTERIURIA TREATMENT A number of studies were done on antibiotic prophylaxis and bacteriuria treatment with individuals with neurogenic bladders. Kass found that if there is no VUR, bacteriuria is innocuous; in his study, 17 hydronephrotic kidneys showed significant radiographic improvement since starting CIC (125). Ottolini found that asymptomatic bacteriuria requires no antibiotic therapy in the absence of VUR (126). Van Hala found that there is no correlation among the number of UTIs, the type of catheter used, and the use of prophylactic antibiotics (127). Johnson and colleagues found that nitrofurantoin is an effective prophylactic agent during a 3-month period for bacteriuria (128). Schlager and colleagues found that asymptomatic bacteriuria persists for weeks in children with neurogenic bladders with normal upper UTs managed with CIC (129). The asymptomatic bacteriuria is different from the symptomatic bacteriuria. Jayawardena and colleagues found that patients with a spinal cord injury (SCI) frequently have asymptomatic bacteriuria without data to support treatment and that routine UCs should not be done at annual evaluations (130). (Note: It may be appropriate for a pediatric patient without a neurogenic bladder and with frequent UTIs secondary to dysfunctional voiding to receive prophylactic antibiotics for a time. Patients with VUR and with or without a neurogenic bladder routinely receive prophylactic antibiotics.) NEONATAL VERSUS CHILDHOOD TREATMENT Early proper management is imperative for the preservation of renal function (131). A urologist should be involved from the newborn period as 20% are born with renal anomalies and kidney damage was found to be approximately 1 in 4 without proper management of the neurogenic bladder (115). Treatment of neurogenic bladder dysfunction due to myelomeningocele in neonates is recommended. A study of 98 individuals (46 started CIC in the first year of life, 52 began CIC after 4 years of life) reviewed the charts of those using CIC who were believed to be at risk for renal deterioration. The mean follow-up of this study was 4.9 years, and the average age of the patient at the last follow-up was 11.9 years. The study found that neonatal treatment enabled UDY to identify those infants at risk for upper tract deterioration, which was prevented by the initiation of oxybutynin and CIC. There was a similar improvement in UTI rate, hydronephrosis, and reflux. The percentage of patients with worsening hydronephrosis and persistent high intravesical pressures who needed bladder augmentation was 11% in the earlier treatment group versus 27% in the later treatment group (P < .05) (132). FURTHER SURGICAL MANAGEMENT FOR REFLUX AND SMALL NEUROGENIC BLADDERS Lowering the bladder pressure below 40 cm H2O is the goal. Ureteral reimplantation may be necessary for reflux; however, most people with neurogenic bladders have reflux from a high-pressure bladder and not from a ureterovesical junction that is dysfunctional. Medical management with CIC and anticholinergic medication frequently causes reflux to resolve, and surgical correction is rarely necessary. Bladder augmentation may be considered for a small or high-pressure bladder refractory to medical treatment. Bladder augmentation may increase the risk of bladder cancer, rupture, and stone, and mucus may be excessive in the urine, obstructing CIC (133). For cancer surveillance in those with augmented bladders, cystostomy was recommended annually starting 10 years following the bladder augmentation (118). Newer recommendations may be different and a urologist should be involved to determine adequate surveillance. Augmentation should be explored only after pharmacologic management has failed and the system continues to be a high-pressure system, thus putting the kidneys at risk. The complication rate for bladder augmentation in one study was approximately 1 in 3 (134). In conclusion, those individuals with a normal neurologic exam with sacral-level spina bifida likely have a neurogenic bladder and need appropriate management. This point is demonstrated in a study of bladder dysfunction and neurologic disability at presentation in closed spina bifida. There were 51 individuals in the study, with a mean age of presentation of 3.3 years. Of these patients, 25 had UT disturbance, 12 had neurologic problems, 33 had a normal neurologic exam, 21 had a normal renal ultrasound (RUS), and 31 had abnormal videourodynamics, despite a normal neurologic exam and RUS (135). The majority of individuals with spina bifida have a neurogenic bowel and bladder; even if there are no motor signs of weakness, infants found to have perineal sensation are likely to be continent as well as have decreased renal complications and improved survival long term (136). The management of neurogenic bladder starts in the NICU in the perinatal period by the urologist, physiatrist, nursing, and family. SOCIAL ASPECTS The bladder focus may put considerable strain on the family (137). Children and adolescents with neuropathic bladders using intermittent catheterization have worries about peers discovering catheters used to empty their bladder and regarding leakage of urine. Urinary incontinence does affect self-esteem, and it is important to aim medical management at continence for psychological (138) as well as physical well-being. Urinary continence is an important developmental milestone in individuals with and without spina bifida (138). As mentioned in the previous edition of this book, urinary continence, although important, should not occur at the expense of the kidneys. NEUROGENIC BOWEL NEUROGENIC BOWEL DYSFUNCTION The colon, rectum, and internal anal sphincter are also controlled by autonomic nerves. Parasympathetic innervation is from S2 to S4, whereas sympathetic fibers arise from the lower thoracic and lumbar segments. Voluntary somatic motor and sensory nerve supply for the external anal sphincter is from S2 to S4 through the pudendal plexus. Coursing through the spinal cord, these nerves have direct connections with the integrating supraspinal centers in the pons and cerebral cortex (56). Colon peristalsis propels feces into the rectum. The gastrocolic reflex increases peristalsis for about 30 minutes after food intake. Rectal fullness initiates an autonomic stretch reflex, with relaxation of the internal anal sphincter, and creates a sensation of predefecation urge. In contrast, the voluntarily controlled external sphincter remains contracted to retain feces. When the situation warrants, this action is further enhanced by voluntary contraction of the levator ani, and gluteal muscles. Defecation occurs when the external sphincter is voluntarily relaxed (56). Many children with spina bifida have a patulous anus, absent cutaneous reflex response, and perianal sensory deficit. This indicates dysfunction of S2 to S4 segments and leads to fecal incontinence. With lesions above L2, there can be an intact reflex arc to maintain perineal sphincter tone, despite absent rectal sensation. BOWEL MANAGEMENT Fecal incontinence and constipation are common problems with spina bifida. The external anal sphincter can be weak and patulous. The reflex arc can be interrupted, causing an interruption of innervation to the anal sphincter. There are also motility issues affecting the entire colon. Bowel management is essential for a successful bladder program in individuals with spina bifida. Not only are gastrointestinal (GI) symptoms and incontinence, with its self-esteem issues, reduced, successful treatment of constipation may improve bladder storage and reduce the frequency of UTIs. Daily bowel movements should be a goal. Aspects of daily bowel care include drinking enough fluids, using an osmotic laxative such as polyethylene glycol, a high-fiber diet to bulk up the stool, digital rectal stimulation or a glycerin suppository in infants and younger children, a bisacodyl suppository 5 mg rectally in younger children and 10 mg rectally in those at least older than 2 years (usually school age for the higher dose). The suppository will not work if placed directly on the stool in the rectum, so careful placement on the rectal mucosal wall is essential. In those with a patulous anus, suppositories may be ineffective because of inadequate ability to retain it. Although the use of the gastrocolic reflex is questionable in the spina bifida population, it is still advised to try having the bowel movement approximately 20 to 30 minutes after the nightly meal. (The morning or afternoon meal are both okay, too, but secondary to schedules, it may be difficult to embark on a bowel program just before or during school or work.) If the bowels are void of stool, incontinence of stool and urine is less likely. With severe constipation seen on an abdominal film, or with palpable stool still in the abdomen, the previous procedure should be followed along with an enema. If the anus is patulous, a cone tip may be needed to administer the enema and keep the fluid within the rectum for a sufficient length of time. Anatomic bowel obstruction should be ruled out by abdominal x-ray in severe constipation before a colonic cleansing enema is performed. A surgical procedure may be necessary, such as a catheterizable appendicocecostomy through the abdominal wall to flush the large intestine from the proximal end with an enema (139). This is known as the ACE (antegrade colonic enema) procedure or MACE procedure (M for Malone who popularized the procedure). The placement of a cecostomy tube is another method to access the bowel for antegrade enema. A successful bowel program maintains bowel continence and can be performed independently or with minimal supervision. Anorectal manometry and biofeedback in the presence of intact or partial rectal sensation anocutaneous reflex offer encouraging results. Rectal sensation is considered normal when a rectal balloon inflated with 10 mL of water or less is perceived. The external sphincter activity can be recorded with surface electrodes. Repeated sessions of inflating and deflating the balloon comprise the biofeedback training (56). This can be done during a urodynamic procedure. For some patients, this will work to see the tracings and surface electromyography (EMG) electrode recordings when attempting to volitionally close the anal sphincter. Social acceptance from peers is important and leakage of stool can be difficult in school and other social situations. Important goals include continence of stool by grade school entry as preparation for independence and increased career options.
 Usually normal neurologic exam
Usually normal neurologic exam
 No association with hydrocephalus or Chiari malformation
No association with hydrocephalus or Chiari malformation
 Uncommon: occurs less than 10%
Uncommon: occurs less than 10%
 Most common
Most common
 Associated with hydrocephalus and Chiari type 2 malformations
Associated with hydrocephalus and Chiari type 2 malformations
 Abnormal motor and sensory exam
Abnormal motor and sensory exam
 Neurogenic bowel and bladder
Neurogenic bowel and bladder
 75% in the lumbosacral segment
75% in the lumbosacral segment
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


