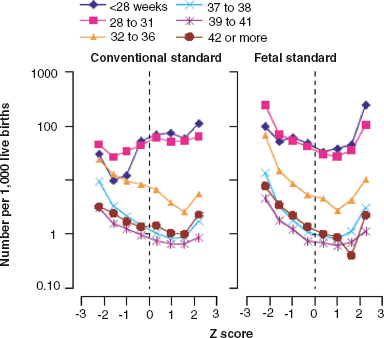
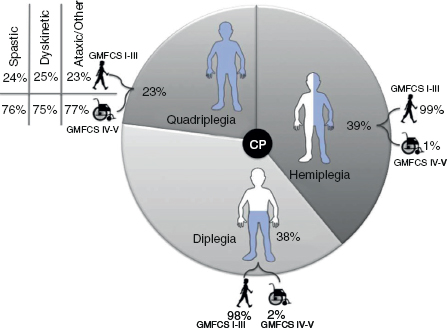
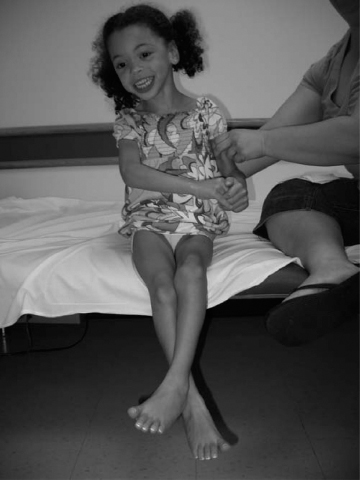
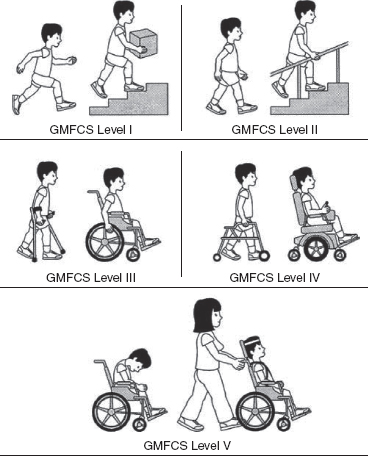
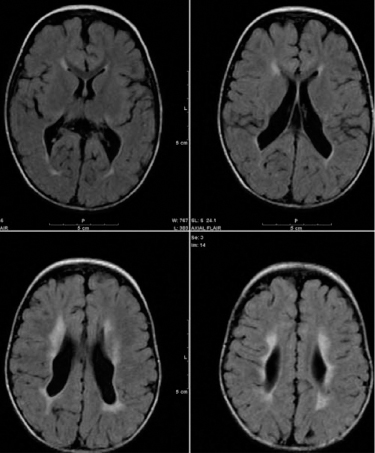
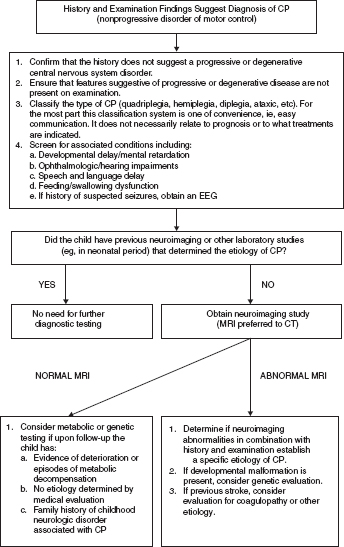
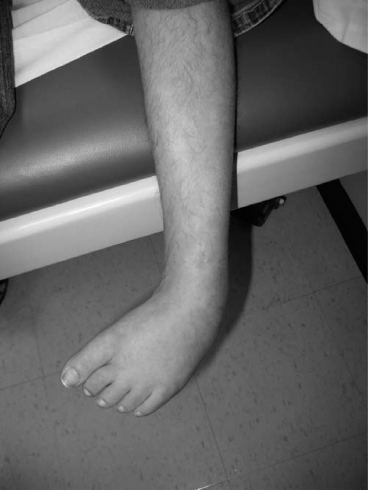
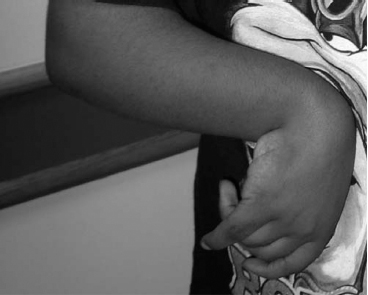
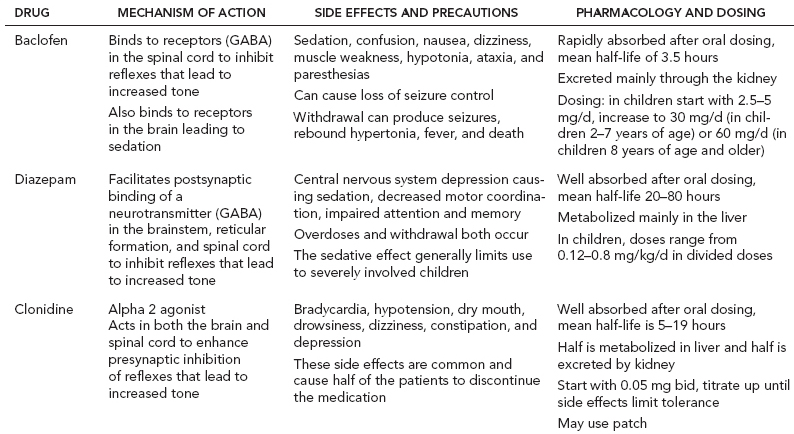
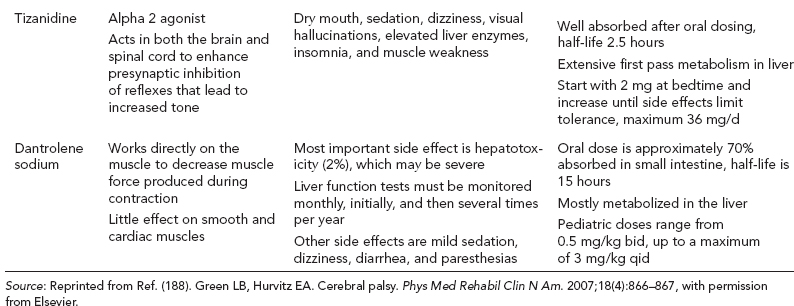
14 CEREBRAL PALSY Mary McMahon, David Pruitt, and Jilda Vargus-Adams Cerebral palsy (CP) is defined as “a group of permanent disorders of the development of movement and posture, causing activity limitations that are attributed to nonprogressive disturbances that occurred in the developing fetal or infant brain.” (1). There are three major criteria for diagnosis of CP: a neuromotor control deficit that alters movement or posture, a static brain lesion, and acquisition of the brain injury either before birth or in the first years of life. Due to the breadth of these criteria, CP is an extremely heterogeneous diagnosis in terms of clinical presentation, etiology, and pathology. Although the brain lesions that result in CP are not progressive, the clinical picture of CP may change with time as the affected individual grows and develops. EPIDEMIOLOGY AND RISK FACTORS CP is the most common motor disability of childhood, affecting approximately 3.1 per 1,000 school-age children (2) with at least 8,000 new cases each year in the United States (3). The population of children with CP may be increasing due to premature infants who are surviving in greater numbers (4), higher incidence in normal-weight term infants (3), and longer survival overall. The proportion of CP that is most severe is also increasing, with as much as a third of all children with CP having both severe motor impairments and intellectual disability (5). The etiology of CP is often not well understood. The majority of cases in term infants do not have a clear etiology, although some risk factors have been identified including placental abruption, birth asphyxia, and neonatal medical problems (6). Factors that may contribute to brain injury and CP include prematurity, infection, inflammation, and coagulopathy (7). There is also considerable interest in the contributory roles of various biomolecules and cytokines that accompany infectious or inflammatory processes (8). Risks such as consanguinity, Rhesus incompatibility, vaccine-preventable infections, and iodine insufficiency are more common in the developing world (9). The greatest risk factor for the development of CP is prematurity. Premature infants (born earlier than 37 weeks’ gestation) are much more likely to develop the condition than term infants, and incidence rates are highest in the very earliest infants (9,10). Rates of CP in premature and low birth-weight infants vary from 40 to 150 per 1,000 live births (11), with some reports suggesting increasing (4) or decreasing rates (11,12) in the past two decades. Figure 14.1 (10) demonstrates the roles of prematurity and birth weight in CP. The vertical axis shows the incidence rates of CP for groups of varying gestational age. Higher rates, approaching 1 in 10 live births, are evident for premature birth of less than 32 weeks’ gestation age whereas term births show rates closer to 1 in 1,000 live births. The horizontal axis shows variation from standard or preferred birth weights as determined by gestation age and represented by z scores. The effect of low (or high) birth weight is demonstrated with the “U”-shaped curves of CP incidence peaks across most gestational ages. This figure demonstrates the profound effect of prematurity as a risk factor for CP. Prenatal risk factors for CP include being small for gestational age (13), being of low or very low birth weight (14), multiple gestation (15), developing infection (especially chorioamnionitis and cytomegalovirus) (16), having evidence of stroke (17), or having neonatal encephalopathy (18). Maternal risk factors for CP include chorioamnionitis (16) or fever during labor, coagulopathy or bleeding (19), placental infarction, and thyroid disease (20). Postnatal risk factors for CP are often related to social disadvantage, and include trauma in developed nations (21) and infection in developing nations (22). Additional risk factors for CP include kernicterus (23), methyl mercury exposure (24), and genetic causes (25). Severe birth asphyxia in term infants is not a major cause of CP. A small minority of children with the condition had asphyxia (6,26), in contrast to prematurity, which is associated with up to half of all cases of CP. Nonetheless, for children who have true birth asphyxia, the risk of CP is increased (27). Fetal monitoring in the United States has probably increased the rate of cesarean section deliveries, but has not been associated with any decline in rates of CP (28). Term infants described as having birth asphyxia often manifest certain signs, including acidosis, bradycardia, or neonatal encephalopathy. Intrauterine exposure to infection or a coagulation disorder can cause a similar clinical picture at birth and may be mistaken for complications of birth asphyxia. Neonatal encephalopathy generally is diagnosed in neonates with significant neurologic dysfunction, including respiratory difficulties, altered tone, low consciousness, and/or seizure activity. It is the best predictor of CP in term infants, regardless of the cause of the encephalopathy. FIGURE 14.1 Prevalence of CP by z score of weight for gestation. Source: Reprinted with permission from Ref. (10). Jarvis S, Glinianaia SV, Torrioli MG, et al. Cerebral palsy and intrauterine growth in single births: European collaborative study. Lancet. 2003;62:1106–1111. CLASSIFICATION CP has traditionally been classified by type of movement disorder and anatomic distribution; see Figure 14.2 (29). FIGURE 14.2 Proportion of CP by topography and severity. Source: Reprinted with permission from Ref. (29). Novak I. Evidence-based diagnosis, health care, and rehabilitation for children with cerebral palsy. J Child Neurol. 2014;29:1141–1156. FIGURE 14.3 A child with dystonic CP. Movement patterns include spastic, dyskinetic, hypotonic, ataxic, and mixed forms. The most common movement pattern is spastic including 77% in the United States (2) with a minority of cases being primarily dyskinetic, ataxic, or hypotonic (30). The distinction between spasticity and dystonia is not always clear. An interdisciplinary group developed a consensus statement on the definition of each term. Spasticity was defined as hypertonia in which one or both of the following signs are present: (a) resistance to externally imposed movement increases with increasing speed of stretch and varies with the direction of joint movement, and/or (b) resistance to externally imposed movement rising rapidly above a threshold speed or joint angle (31). Dystonia was defined as a movement disorder in which involuntary sustained or intermittent muscle contractions cause twisting and repetitive movements, abnormal postures, or both (Figure 14.3) (31). Hypotonic and ataxic forms of CP are rare and, therefore, any child suspected of having either of these diagnoses should receive a thorough diagnostic evaluation for other neurologic conditions. FIGURE 14.4 A child with hemiparetic CP. The anatomic distribution of motor problems in CP is the primary means of classification. The three categories of hemiparesis, diparesis, and quadriparesis occur with fairly equal frequency (5,30). Hemiparetic CP affects only one side of the body and typically demonstrates greater impairments in the upper extremity (UE; Figure 14.4). Diparetic CP affects the lower extremities more than the upper extremities (Figure 14.5). Quadriparetic CP affects the entire body, including the axial as well as appendicular skeleton (Figure 14.6). An interest in classifying children with CP based on function in addition to the distribution of motor impairment resulted in the development of the Gross Motor Function Classification System (GMFCS). The GMFCS stratifies children with CP into five groups based on gross motor skills (32); see Figure 14.7. In this system, specific descriptions of mobility functions, based on age, allow each child with CP to be categorized. In GMFCS I, children walk indoors and outdoors and climb stairs without limitation. Children who are GMFCS II walk indoors and outdoors and climb stairs holding onto a railing but experience limitations walking on uneven surfaces and inclines. Children who are GMFCS III walk indoors or outdoors on a level surface with an assistive mobility device. Children may climb stairs with a railing or propel a manual wheelchair. Children who are GMFCS IV may walk short distances with a device, but rely more on wheeled mobility at home and in the community. Children at GMFCS V have no means of independent mobility. A related classification system for UE function, the Manual Abilities Classification System, permits categorization by fine motor performance (33), and the Communication Function Classification System applies to language and communication, including cognitive and motor performance (34). FIGURE 14.5 A child with diparetic CP. FIGURE 14.6 A child with quadriparetic CP. PATHOLOGY More than 80% of children with CP will have abnormal findings on neuroimaging (35–37). These abnormal findings can provide valuable clues to pathogenesis. FIGURE 14.7 The Gross Motor Classification System for children aged 6 to 12 years. Source: Reprinted with permission from Graham HK. Classifying cerebral palsy. J Pediatric Orthop. 2005;25:128. The most common abnormality on neuroimaging is found in the white matter near the lateral ventricles and is called white matter disease of prematurity. Findings may include various lesions, the most significant of which is periventricular leukomalacia (PVL) (38), with reports of up to 56% of all cases of CP demonstrating abnormalities in this location (37) (Figure 14.8). PVL occurs much more commonly in premature infants than in term infants (90% vs 20%), often consistent with an insult in the second trimester, and is a common outcome of intraventricular hemorrhage in premature infants (37). Because the corticospinal tract fibers to the lower extremities are medial to those of the upper extremities in the periventricular white matter, children with PVL typically have spastic diparesis. One large study found that PVL was present in 71% of the children with diparesis, 34% of those with hemiparesis, and 35% of those with quadriparesis (35). Deep gray matter lesions to the basal ganglia and thalamic region are mainly associated with dystonic CP, and have been found in approximately 12% of children with the condition (35). Historically, large numbers of children acquired athetoid CP following a diagnosis of kernicterus, due to concentrated damage to the basal ganglia with bilirubin encephalopathy. These cases are far less common with advancements in the treatment of neonatal jaundice. FIGURE 14.8 Periventricular leukomalacia. Focal cortical infarcts involving both the gray and white matter are found almost exclusively in patients with hemiparesis, and are typically related to middle cerebral artery strokes. In a group of children with hemiparetic CP, 27% were found to have a focal infarct on imaging (35). Brain malformations can be found on neuroimaging in approximately 10% of children with CP (35–37,39). Neuronal migration disorders or insults early in pregnancy can result in lissencephaly, polymicrogyria, schizencephaly, or holoprosencephaly. Some in utero infections, such as those caused by cytomegalovirus, can also cause distinctive brain malformations (35). Brain malformations are more commonly found in cases of term infants and hemiparesis (36). Children who sustain diffuse brain insults demonstrate more extensive injury on neuroimaging. Infection and ischemia are two of the more common causes of generalized encephalomalacia. A wide range of findings may be present on MRI, including multiple cysts, cortical thinning, white and gray matter damage or loss, and microcephaly. Children with diffuse brain lesions or anomalies typically demonstrate spastic quadriparesis and are at high risk for additional medical and cognitive problems. INITIAL EVALUATION AND CLINICAL FINDINGS SIGNS AND SYMPTOMS Early identification of children who have CP allows for early therapeutic intervention and screening for associated conditions. Because CP is a descriptive term that does not infer a single etiology, pathology, or prognosis, there is no specific diagnostic test. It is a diagnosis of exclusion based on a careful history and physical exam. It can be difficult to make a definitive diagnosis in infants less than 6 months old. Prior to this time, the infant has a limited repertoire of volitional movements, which makes milder delays in motor development difficult to detect. In addition, abnormalities in tone and reflexes are often subtle in early infancy. As the cortex matures in the second half of the first year, the diagnosis typically becomes more apparent. The first step in the evaluation for suspected CP is a comprehensive history, including a detailed account of potential risk factors and family history. A thorough history of developmental milestones is also important. Often the parent’s initial concern is a significant delay in attaining motor milestones. Prematurity must be considered when evaluating development because milestones are generally corrected for the degree of prematurity. A discrepancy between motor and cognitive milestones should always raise suspicion for CP. Certain deviations in developmental milestones are associated with CP. For example, early hand preference or asymmetric use of the extremities may be the first indication of hemiparesis. Early head control, rolling, or rigid standing are all associated with abnormally increased tone and/or exaggerated primitive reflexes. The parent may also describe unusual means of mobility, such as bunny hopping, combat crawling, or bottom scooting. The most important aspect of the developmental history is to confirm that the child has not lost any skills or milestones, as this would suggest a neurodegenerative disorder. Following a detailed history, a thorough physical examination should be performed. A careful neurologic exam is an essential piece of the evaluation. In infancy, the neurologic exam focuses on tone and infantile developmental reflexes. Deep tendon reflexes, plantar responses, and the presence of clonus are more informative in the older child. Tone should be assessed by gently moving the infant’s joints through their appropriate range of motion (ROM) and evaluating the amount of resistance. Careful observation will also provide information about an infant’s tone. Infants with severe hypotonia will lay in a frog-leg position with their hips abducted, flexed, and externally rotated. Their arms will lie limply at their sides. Persistent fisting or scissoring may be observed with increased tone. Most infants will undergo an early stage of mild or moderate hypotonia prior to more traditional signs of CP. A prolonged period of hypotonia or fluctuating tone is more typical of dyskinetic CP. In general, however, longer periods of hypotonia and severe hypotonia are associated with more severe motor deficits, regardless of the type of CP. The earliest indication of CP may be a delay in the disappearance of primitive infantile reflexes. Commonly examined primitive reflexes include the Moro reflex, palmar grasp reflex, asymmetric tonic neck reflex, and tonic labyrinthine reflex. During the first 6 months of life, maturation of the cortex gradually overrides these primitive responses, and voluntary motor activity should increase. Persistence of these primitive reflexes past 6 months of age, asymmetry of the response, or an obligatory response at any age should be considered highly suspicious for a significant motor impairment. As the primitive reflexes become suppressed, postural or protective reactions such as the parachute and the equilibrium or tilting reactions should emerge. In children with CP, postural reactions may be less effective, appear later than usual, or fail to develop. A general movement assessment (GMA) can also be useful in evaluating a child for CP. A systematic review of tests, including GMA, neurologic examination, MRI, and cranial ultrasound, found that the GMA had the best evidence and strength for predictive accuracy in high-risk infants (40). GMA involves observation of the infant’s spontaneous movements. Abnormal general movements in a young infant are described in the following three categories: (a) poor repertoire: monotonous movement sequences, (b) cramped synchronized: muscles contract and relax almost simultaneously, resulting in the movements being rigid, (c) chaotic: large-amplitude movements of all limbs that lack fluency or smoothness (41). At 3 months’ corrected age, general movements of a fidgety nature are typical. Normal fidgety movements included small movements of moderate speed and variable acceleration of neck, trunk, and limbs in all directions that are continual in the awake infant except during fussing and crying. Abnormal fidgety movements are of larger amplitude with moderately exaggerated speed and jerkiness (41). A systemic review found that the pooled sensitivity and specificity for GMA in predicting CP were 98% and 91% respectively (40). Formal assessment tools, such as the Developmental Assessment of Young Children (42) and the Hammersmith Infant Neurological Evaluation (43) have also been shown to be highly predictive of CP in infants less than 12 months of age. IMAGING Neuroimaging can be helpful in determining the etiology of CP and the timing of the insult. The Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society published two practice parameters that address the use of neuroimaging in the neonate and the child with suspected CP (44,45) (Figure 14.9). Recommendations for imaging in the preterm neonate include a screening cranial ultrasonography on all infants with less than 30 weeks’ gestation, between 7 and 14 days of age, and again between 36 and 40 weeks’ postmenstrual age (44). This recommendation was based, in part, on the fact that a 10-fold elevation in the risk of adverse outcome for the very low birth-weight infant was identified with ultrasound evidence of grade 3 or 4 intraventricular hemorrhage, periventricular cystic lesions, or moderate to severe ventriculomegaly (44). In term infants with neonatal encephalopathy, the practice parameter recommends a noncontrast computed tomography (CT) to detect hemorrhagic lesions when there is a history of birth trauma, low hematocrit, or coagulopathy (44). If the CT is inconclusive, MRI should be performed between days of life 2 and 8 in order to assess the location and extent of injury. Abnormalities of the thalamus and basal ganglia were associated with increased neurodevelopmental disability at 1 to 2 years of age (44). Neuroimaging can also be useful in determining an etiology in children suspected of having CP outside of the neonatal period. The practice parameter on the diagnostic assessment of the child with CP found an abnormal MRI scan in the majority of children with CP (average 89%) and that MRI was more likely to show an abnormality when compared to CT (average 77%) (45). The practice parameter, therefore, recommends neuroimaging in the evaluation of a child with CP if the etiology has not been established, and MRI is preferred to CT. LABORATORY FINDINGS Metabolic or genetic causes for CP are unusual, and laboratory studies to investigate these conditions are not routinely recommended. Metabolic or genetic testing is recommended in the following conditions: if neuroimaging does not determine a specific structural abnormality or if it reveals a developmental malformation, if there is evidence of developmental deterioration, or if there is a family history of a childhood neurologic disorder associated with a diagnosis of “cerebral palsy” (45). The practice parameter also recommends consideration of diagnostic testing for a coagulation disorder in children with an unexplained cerebral infarction on neuroimaging (see Figure 14.9). DIFFERENTIAL DIAGNOSIS Young infants with CP often present with hypotonia. The differential diagnosis for the floppy infant is vast. The most common etiologies include central nervous system disorders such as CP, neuromuscular disorders, genetic disorders, and metabolic disorders. Clues to a neuromuscular disorder include diminished deep tendon reflexes, weakness (which may result in absent infantile reflexes), or a positive family history. Dysmorphic features may suggest a genetic cause for hypotonia, such as Down syndrome, Prader–Willi syndrome, or Angelman syndrome. Metabolic disorders may present at any age, but are most likely to present in infancy. Metabolic disorders should be considered if a previously healthy child presents with an acute encephalopathy without an adequate explanation. Metabolic acidosis, hypoglycemia, hepatic involvement, or cardiac involvement should also prompt consideration of a metabolic disease. Dystonia and spasticity are present in a number of metabolic disorders, including mitochondrial disorders, glutaric aciduria type I, Lesch–Nyhan syndrome, and homocystinuria. A diagnosis other than CP should always be sought in children who have evidence of progressive disease or loss of previously obtained milestones. FIGURE 14.9 Algorithm for the evaluation of the child with CP. Source: Reprinted with permission from Aswal S, et al. Practice parameter: Diagnostic assessment of the child with cerebral palsy. Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2004;62:851–863. A definitive diagnosis of CP should be made cautiously, especially in the first 6 months of life. Infants who are suspected of having CP should be followed closely with serial developmental evaluations and physical exams until the diagnosis is clear. An early diagnosis is considered best practice, however, because it will allow access to diagnosis-specific early intervention and parental support (29). ASSOCIATED DISORDERS SENSORY IMPAIRMENTS CP is defined as a disorder of movement control and posture, and therefore sensory impairments are easily overlooked. Deficits in two-point discrimination, proprioception, and stereognosis have been described (46–48). Sensory deficits are believed to be most common in children with hemiparesis. A study of children with spastic hemiparesis found that 97% of the spastic limbs had a stereognosis deficit, 90% had a two-point discrimination deficit, and 46% had a proprioception deficit, and these sensory deficits were more commonly present in limbs with a greater size discrepancy (48). Sensory deficits can also be found in the limbs that do not appear to be affected by CP (49). Bilateral sensory deficits were found in 88.8% of children with hemiparesis in one study (46). Stereognosis and proprioception deficits were the most common bilateral abnormalities, and the extent of sensory loss did not mirror the motor deficit. Another study identified abnormalities of tactile spatial discrimination in the hands of children with spastic diparesis with apparent normal motor function in their upper extremities (47). Little is known about the significance of right- or left-sided brain injury on sensory deficits in CP. A trend toward children with left hemiparesis to perform worse on spatial-tactile tests has been reported (49). Beyond the brain injury itself, it has been hypothesized that a reduced number of perceptual motor experiences throughout development may contribute to the reorganization of the sensorimotor cortex (50). Sensory deficits are important to recognize because they can significantly affect functional use of the extremity. VISUAL IMPAIRMENTS Visual impairments are common in children with CP, with a reported prevalence of 39% to 100% (51). The inherent difficulty in doing an ophthalmologic exam on children with varying degrees of cognitive and motor impairments makes it difficult to determine the precise incidence of visual disorders. Strabismus is the most commonly reported visual disorder, but a wide variety of other disorders have been described. There is emerging evidence that each clinical type of CP may be associated with a distinct neuro-ophthalmologic profile. In a study of 129 patients with CP who underwent an extensive evaluation of the visual system, investigators found that visual dysfunction in diplegia was characterized mainly by refractive errors, strabismus, abnormal saccadic movements, and reduced visual acuity (52). The participants with hemiplegia showed strabismus and refractive errors while oculomotor involvement was less frequent. Altered visual field was most common in subjects with hemiplegia. Children with tetraplegia had the highest rate of visual impairment, characterized by ocular abnormalities, oculomotor dysfunction, and reduced visual acuity (52). Another study demonstrated a relationship between visual deficits and severity of CP as measured by the GMFCS (53). In this study, children in each level of the GMFCS had visual deficits 10- to 70-fold higher than those reported in the general age-matched pediatric population. Children with milder CP, GMFCS levels I to II, had visual deficits that resembled neurologically normal children with strabismus and amblyopia. Children with the most severe CP were at greatest risk for high myopia, absence of binocular fusion, dyskinetic strabismus, severe gaze dysfunction, and optic neuropathy or cortical visual impairment (53). Visual impairments likely play a pivotal role in motor and cognitive function in children with CP (52). Early assessment and accurate detection of specific disorders is an important aspect of the rehabilitation care and overall quality of life (QOL) of these children. HEARING IMPAIRMENTS Considerable variation exists in estimates of hearing loss in CP, in part due to the difficulty in assessing hearing, as well as the population studied. A systematic review of population-based data on children with CP revealed a range of reported hearing loss of 4% to 13% and a range of severe hearing loss of 2% to 12% (54). The proportion of severe/profound hearing loss appears to be increasing in CP and this is felt to be related to the increasing number of survivors of extremely preterm birth or better detection (54). Sensorineural hearing loss is most commonly associated with congenital toxoplasmosis, rubella, cytomegalovirus, and herpes (TORCH) infections, bacterial meningitis, hypoxia, and ototoxic drugs. In the past, kernicterus was a relatively common cause of sensorineural hearing loss in athetoid CP. Hearing loss can significantly impact the development of communication and cognition and therefore early assessment and accurate detection are recommended. COGNITIVE IMPAIRMENTS Cognitive impairments are common in CP. It is difficult to make generalizations about the specific relationship of CP and cognitive function because CP is a heterogeneous disorder and the available literature often does not differentiate between the various types. In addition, assessment of intellectual functioning can be difficult in patients with severe motor and communication difficulties, which may lead to an underestimation of cognitive function. An overestimation of cognitive function can occur in patients who are socially responsive. The overall frequency of an IQ score of 69 or below is reported to be 50% to 70% (55). In general, patients with more severe neuromuscular impairments are at greater risk for cognitive impairments, but some patients with severe motor impairments can have normal cognition. For example, a patient with athetosis secondary to a discrete lesion in the basal ganglion is likely to have normal intelligence. Recent research has suggested that disorders of higher level cognitive tasks and executive functioning (EF) are also common in CP (56–58). Even children with mild unilateral CP can have significant EF impairments in comparison to typically developing children (56). A systematic review of brain structure and EF in CP found that parieto-occipital periventricular hemorrhage, intraventricular hemorrhage, ventricular dilatation, white matter reduction and bilateral lesions were associated with greater EF impairments (59), but in general there is a paucity of brain imaging studies focused on EF in children with CP. It is important to identify EF dysfunction early on due to its effect on academic and vocational success, as well as social integration. Therefore a systematic neuropsychological examination with particular attention to EF should be obtained in children with CP (60). EMOTIONAL AND BEHAVIORAL IMPAIRMENTS The prevalence of emotional and behavioral problems in different populations of children with CP is reportedly 30% to 80% (61) but in general, it has not been well defined in the literature. A wide variety of behavioral and emotional disorders are possible, including attention deficit disorder, passivity, immaturity, anger, sadness, impulsivity, emotional lability, low self-esteem, and anxiety. A population-based analysis of behavior problems in children with CP identified problem behaviors in 25% of the children as assessed by parent reports (61). Specific behaviors that were most common in this population included dependency, being headstrong, and hyperactivity. An additional population-based study in Europe found a similar prevalence of significant emotional and behavioral symptoms in 26% of children with CP (62). The most common problems identified were in peer relationships (32%), hyperactivity (31%), and emotion (29%). Difficulty with peer relationships has been found even in children with milder CP (GMFCS I). Compared to their classmates, children with mild CP were found to have fewer reciprocated friendships, fewer sociable and leadership behaviors, and were more isolated and victimized by their classmates (63). A potentially important contributing factor to impaired behavioral and social functioning is decreased cognitive EF (64). Although, there is widespread agreement that emotional and behavioral dysfunction is common in children with CP, there is very limited research on effective interventions to address the issue. There is emerging evidence that parenting interventions and cognitive behavioral therapy, such as the Stepping Stones Triple P and Acceptance and Commitment Therapy, may be beneficial (65). An early awareness of the risk for emotional and behavioral disorders by professionals and parents is important, so that they can be recognized early. Referral to a mental health specialist should be considered, as well as a neuropsychological evaluation, including EF. EPILEPSY The overall occurrence of epilepsy is reported to be between 15% and 55% in a mixed population of children and adults with CP (66). A wide variety of types of seizures are possible, and a clear correlation between various risk factors and seizure frequency or type has yet to be established. Seizures are more common in children with more severe CP and in children with quadriparesis and hemiparesis versus diparesis (67). OROMOTOR IMPAIRMENTS Oromotor impairments are associated with more severe CP. A weak suck, poor coordination of the swallowing mechanism, tongue thrusting, and a tonic bite reflex may all lead to feeding difficulties and increased risk for aspiration. Speech disorders range from mild articulation disorders to anarthria, and are most commonly seen in children with spastic quadriparesis or athetosis. Oromotor dysfunction may also lead to difficulty controlling oral secretions and drooling, which may negatively affect social interactions. A wide range of interventions exist to address drooling, but a recent Cochrane review concluded that there is insufficient evidence to inform clinical practice (68). Oromotor impairments are also associated with dental malocclusion and difficulty with oral hygiene, leading to an increased risk of periodontal disease. NUTRITIONAL DISORDERS The assessment of growth and nutrition in children with CP can be difficult due to the lack of a reliable means of measuring stature in children with contractures and scoliosis and the lack of appropriate reference data or growth curves specific to CP (69). Population-based growth patterns of CP have been published (70), but they probably include many children with conditions affecting growth and feeding, and therefore should not be considered prescriptive of how children with CP should grow (69). For children who are unable to obtain adequate nutrition by mouth, gastrostomy or jejunostomy tubes are often utilized. There is evidence that this results in improved weight gain, but there is the potential for adverse events including overfeeding, site infection, stomach ulcer and reflux (73). A recent Cochrane review of gastrostomy feeding versus oral feeding alone in children with CP did not reveal a single randomized controlled trial (RCT) on this topic (74). Given the lack of evidence to guide clinical practice, the decision on the most appropriate means of nutritional support should be made in the context of each individual patient and family. Although malnutrition is a primary concern, children with CP are also at risk for overfeeding and obesity. Children with more severe CP have lower total energy expenditure and higher body fat content than age- and sex-matched children without disabilities, placing them at risk for overfeeding with energy-dense enteral feeds (75). A study of ambulatory children with CP showed an increase in the prevalence of obesity from 7.7% to 16.5% over a 10-year period, an increase similar to that seen in the general pediatric population in the United States (76). GENITOURINARY DISORDERS The development of urinary continence is typically delayed in children with CP. A study of 601 children with CP found that by the age of 6, 54% of children with spastic quadriparesis and 80% with spastic hemiparesis or diparesis had gained urinary continence spontaneously (77). The most important factors associated with urinary incontinence were quadriparesis and impaired cognition. Incontinence was the most common complaint, but frequency, urgency, hesitancy, and urinary retention may also be present (77). Frequency and urgency are often associated with spasticity of the detrusor muscle, causing small, frequent voids. Detrusor over activity and a small bladder capacity were the most common findings on urodynamic studies in children referred for voiding dysfunction, but a minority of children were also found to have detrusor sphincter dyssynergia (78,79). The association between lower urinary tract dysfunction and upper urinary tract dysfunction in CP is unclear. In general, it is reported to be uncommon (80). Symptoms of detrusor sphincter dyssynergia (interrupted voiding, urinary retention, and hesitancy) and culture proven febrile urinary tract infections were shown to correlate with upper tract dysfunction in one study (81). Urodynamic studies should be considered to evaluate the physiology of the bladder in patients with lower tract symptoms. The results can aid in promoting continence, but it is also important to assess basic communication, mobility, equipment and environmental supports, which have also been shown to be important components to obtaining continence (82). RESPIRATORY DISORDERS Children with CP are at increased risk for respiratory illnesses. Impaired control of respiratory muscles, ineffective cough, aspiration due to impaired swallowing, gastroesophageal reflux, and seizures all increase the risk for chronically increased airway secretions. Increased airway secretions may lead to wheezing, atelectasis, recurrent aspiration pneumonia, restrictive lung disease, or bronchiectasis. Bronchopulmonary dysplasia, in an infant born prematurely, will also increase the risk for respiratory disorders. BONE MINERAL DENSITY DISORDERS Decreased bone mineral density (BMD) and increased risk of fracture are present in patients with moderate to severe CP, especially those who are nonambulatory (83). A population-based study revealed that children in GMFCS levels I to III had a similar incidence and pattern for fractures as normally developing children (83). By the age of 10 years, most nonambulatory children have osteopenia, as defined by a BMD z score of less than -2.0 in their femurs (84). Data from the NAGCPP revealed that increasing severity of neurologic impairment, increasing difficulty feeding the child, use of anticonvulsants, and lower triceps skin fold z scores all independently contribute to lower BMD z scores in the femur (84). A published systematic review of available research, used to inform evidence-based clinical practice, concluded that there was probable evidence for bisphosphonates, possible evidence for vitamin D and calcium, and insufficient evidence for weight-bearing activities as effective interventions to improve low BMD. The evidence addressing fragility fractures was sparse. There was possible evidence that bisphosphonates prevent fragility fractures and inadequate evidence to support the use of weight-bearing or vitamin D or calcium to decrease fragility fractures (85). The current evidence for weight-bearing activities is conflicting and consists of studies with small sample sizes, which may result in inadequate power, and studies of relatively short duration and significant variability in how much weight-bearing actually occurred (85). An observational, population study showed a fourfold reduction of nontrauma-related fractures in children who used standers versus those who did not, but this outcome may be attributed to reasons the children were not using standers, such as severe contractures (83). A recent small study demonstrated greater increases in BMD in dynamic loading interventions versus passive, suggesting this method of weight-bearing deserves further study (86). Given the possible effectiveness of vitamin D and calcium supplementation, and the relative safety of this intervention, vitamin D monitoring and supplementation, as well as ensuring adequate calcium intake is recommended for children with CP at risk for osteoporosis (85). MUSCULOSKELETAL DISORDERS Foot/Ankle Foot deformities in CP are typically caused by an imbalance of the muscles that control segmental foot and ankle alignment. This imbalance may be a consequence of spasticity, disrupted motor control, or impaired balance function (87). Equinus deformity, due to increased tone or contractures of the gastrocsoleus complex, is the most common musculoskeletal deformity in CP. Equinovarus foot deformity is primarily due to a combination of spasticity of the posterior tibialis muscle and the gastrocsoleus complex, resulting in inversion and supination of the foot and a tight heel cord (Figure 14.10). This deformity is most common in a child with hemiparesis. In children with diplegia, the varus will often overcorrect into valgus (88). Equinovalgus foot deformity is due to spasticity of the gastrocsoleus complex and the peroneal muscles, as well as weakness in the posterior tibialis muscle. This deformity is most common in older children with spastic diparesis and quadriparesis. Planovalgus foot deformities begin with the lateral displacement of the navicular, causing the talar head to become uncovered and prominent in the mid-foot (88). Hallux valgus deformities are associated with valgus deformities of the foot, which may lead to a painful bunion at the head of the first metatarsal. Knee Knee flexion contractures are common due to spasticity in the hamstring muscles and static positioning in a seated position. If a severe knee flexion is present, hip flexion will be limited, resulting in lumbar kyphosis in the seated position. Flexion contractures at the knee are associated with hip and ankle flexion contractures and patella alta. Genu valgus may also occur, and is most commonly associated with excess femoral anteversion. FIGURE 14.10 Equinovarus foot in a child with CP. Hip Acquired hip dysplasia is common in CP and often leads to progressive subluxation and possible dislocation. Hip subluxation can begin as early as age 2 years (89) and should be monitored closely by exam and serial radiographs. On exam, passive hip abduction of less than 35 degrees and a hip flexion contracture of more than 20 degrees are concerning signs of hip instability (90). On x-ray, hip subluxation is typically defined as a migration percentage greater than 30%. Close surveillance of hip migration with intermittent serial hip radiographs is recommended once hips have subluxed (91). Details of consensus-based hip surveillance programs have been published and include a baseline anterior–posterior (AP) pelvic radiograph at ages 12 to 24 months for all children with CP (92). The reported incidence of dislocation in untreated hips varies, but 25% to 35% is the average estimate from most large series (91,93). Causative factors include persistent excessive femoral anteversion, a dysplastic acetabulum, and muscle imbalance from overactive hip adductors and flexors. These factors cause the hip to be adducted, flexed, and internally rotated, placing it at risk for posterior dislocation. A large population-based sample of children revealed a linear relationship between the incidence of hip displacement and level of gross motor function on the GMFCS (91). The incidence of hip displacement for each GMFCS level was as follows: I 0%, II 15%, III 41%, IV 69%, and V 90%. The natural history of hip dislocation has not been well described. Early osteoarthritis and difficulty with positioning and hygiene are not uncommon. The reported incidence of pain associated with a dislocated hip varies, but is commonly felt to be present in at least 50% of patients with dislocations (93). Children with CP may also develop a “windswept deformity” of their hips, described as an adduction deformity of the elevated hip and an abduction deformity of the opposite hip, which also tends to be externally rotated and commonly results in pelvic obliquity (Figure 14.11). The hip on the elevated side is at significant risk for dislocation, and positioning can be challenging. Hip dislocation with pelvic obliquity is often associated with scoliosis, but any potential causative relationship remains unproven. FIGURE 14.11 Windswept hip deformity in a child with CP. Spine Spinal deformities, including kyphosis, lordosis, or scoliosis, are common in children with CP. Kyphosis is often seen in conjunction with significant weakness of the spinal extensor muscles and tightness in the hamstrings, leading to a posterior pelvic tilt. Lordosis is frequently associated with hip flexion contractures. The likelihood of scoliosis increases with the severity of CP. An overall incidence of approximately 20% (94) has been reported, with an incidence as high as 68% in children with spastic quadriparesis (95). Curves greater than 40 degrees tend to progress, regardless of the patient’s skeletal maturity (95). The risk of progression is greatest for patients with quadriparesis, increased spasticity, a larger curve, a younger age, poor sitting balance, or pelvic obliquity (90). Upper Extremity Spasticity and muscle imbalances can often lead to joint deformities in the UE. The shoulder is often positioned in an adducted and internally rotated position. Spasticity in the biceps, brachioradialis, and the brachialis frequently result in elbow flexion contractures. Elbow flexion contractures less than 30 degrees rarely have functional significance. Forearm pronation deformities are common and can significantly affect functional use of the hand. The most common deformity of the wrist is flexion, typically with ulnar deviation (Figure 14.12). The most common finger deformities are flexion and swan neck deformities due to hand intrinsic muscle spasticity. A thumb-in-palm deformity is commonly seen with adduction at the carpometacarpal joint, which may be associated with hyperextension of the metacarpophalangeal and interphalangeal joints. FIGURE 14.12 Wrist and finger flexion and ulnar deviation in a child with CP. A wide variety of gait classification systems have been developed to assist in diagnosis and clinical decision making, and to facilitate communication among health care providers. A systematic review of the literature, however, concluded that no single classification system appeared to reliably and validly describe the full magnitude or range of gait deviations in CP (96). The following is a description of the more common gait deviations associated with CP (Table 14.1). At the hip, increased hip adduction tone can cause scissoring and difficulty advancing the limb in swing phase. Increased tone in the iliopsoas can lead to increased hip flexion, resulting in an anterior pelvic tilt and a crouched gait. Increased femoral anteversion can contribute to in-toeing. At the knee, tight hamstrings can inhibit the knee from extending during stance phase, further contributing to a crouched gait. Spasticity of the rectus femoris may limit knee flexion during the swing phase, causing a stiff-knee gait pattern. At the ankle, spasticity of the plantarflexors can lead to toe walking, difficulty clearing the foot during swing phase, or genu recurvatum (due to limited dorsiflexion in stance phase creating an extension moment at the knee). Spasticity of the ankle invertors, most commonly seen in spastic hemiparesis, can lead to supination of the foot and weight-bearing on the lateral border of the foot. Weight-bearing on the talar head is more common in spastic diparesis or quadriparesis, and is associated with an equinovalgus deformity. Malrotation of the leg can interfere with stability during stance phase and effective pushoff. Internal rotation is more common with a varus deformity and external rotation with a valgus deformity. TREATMENT GENERAL PRINCIPLES The treatment of a child with CP requires a multidisciplinary approach. Once the diagnosis is made, the infant or child should be evaluated by a comprehensive rehabilitation team. The members of this team will vary, depending upon site and availability. Potential team members may include a physiatrist, developmental pediatrician, orthopedist, neurologist, physical therapist, occupational therapist, speech and language pathologist, therapeutic recreation specialist, orthotist, psychologist, social worker, and a nutritionist. The team should work with the child’s caregivers to develop short- and long-term goals that address neuromuscular concerns such as maintaining ROM and tone control, as well as functional goals related to self-care skills, mobility, and communication. Goals related to increased societal participation should also be included. Goals should be routinely reassessed to ensure that they continue to be valid as the child grows older, and the child should be encouraged to take an active role in goal setting when appropriate. Once the goals are determined, the family and the team must determine the most appropriate therapeutic approach. Although there are many treatment options to choose from, little scientific evidence exists on which to base one’s treatment decisions. The heterogeneity of CP, in addition to the lack of controls and disease-specific outcome measures, all contribute to this lack of evidence. In general, treatment should always start with the least invasive means with consideration of the cost-effectiveness of treatment options. TABLE 14.1 COMMON GAIT DEVIATIONS IN CP LOCATION IMPAIRMENT POTENTIAL EFFECTS Hip Increased adductor tone Scissoring; difficulty advancing leg in swing phase Increased iliopsoas tone Anterior pelvic tilt; increased lumbar lordosis; crouched gait Increased femoral anteversion In-toeing; false genu valgus; compensatory external tibial torsion Abductor weakness Trendelenburg’s gait Knee Decreased hamstring range of motion Crouched gait Hamstring/quadriceps cocontraction Stiff-knee gait Ankle Increased gastrocsoleus tone or contracture Toe walking; genu recurvatum; difficulty clearing foot during swing Internal tibial torsion In-toeing; ineffective pushoff External tibial torsion Out-toeing; ineffective pushoff Varus Increased ankle supination in stance or swing Valgus Increased pronation in stance or swing; mid-foot break PHYSICAL AND OCCUPATIONAL THERAPY Therapy Methods Physical therapists and occupational therapists working with children with CP may choose from a variety of therapy methods, including neurodevelopmental therapy, Vojta, and Rood. There is, however, no clear scientific evidence to support the superior effectiveness of any one particular approach. These methods are sometimes referred to as the neurophysiologic approach, which elicit and attempt to establish normal patterns of movement through controlled sensorimotor experiences (97). This approach focuses on changing factors within the child at the domain of body function and structure, with the assumption that these changes will positively impact the domains of activity and participation. In recent years, the effectiveness of this approach has been questioned (97–99). Some have questioned whether facilitated automatic movement will improve voluntary, active movement (97). This has led to child-active or functional therapy approaches. This therapeutic approach involves actively practicing real-life tasks, typically in real-life environments, for the purpose of gaining skills that are important to the child and the family (29). Proponents of this type of therapy suggest that this approach is most likely to induce maximal neuroplasticity (29). Scientific evaluation of this method is still quite limited, but the level of evidence is highest for this approach in improving hand function (98). An RCT of child-focused therapy (therapy focused on changing identified constraints in the task or environment) versus context-focused therapy (therapy focused on minimizing identified impairments in the child), involving 128 young children with CP, found significant improvements in both groups and no difference in outcomes between groups (100). This suggests that both approaches can be successful. Often, therapists will use a combination of these therapeutic methods. The ideal duration and frequency of therapeutic programs remain unclear. Stretching Children with CP are at significant risk for contracture formation due to muscle imbalances and static positioning. Contractures can interfere with comfortable positioning, functional activities, and care needs, such as dressing, bathing, and toileting. After an initial assessment of baseline ROM, institution of a daily home exercise program with repetitive stretching exercises is usually recommended, although there is no clear evidence to support its efficacy or provide guidance in regard to the ideal frequency or duration (101). There is some evidence to suggest that a sustained stretch is preferable to manual stretching (102). Positioning techniques, orthotic devices, splints, and casting are often recommended to provide a more prolonged stretch. Serial casting is a technique where a series of successive casts are applied in the hope of progressively increasing the ROM with each cast. It is used most frequently at the ankle joint, often in conjunction with botulinum toxin serotype A (BoNT-A), in order to improve dorsiflexion ROM. A systematic review of the literature supports the use of casting at the ankle (98). The evidence suggests a short-term effect on improved ROM (103) and stride length during ambulation (104). Although a number of small RCTs have compared BoNT-A and casting, there is no strong and consistent evidence that casting, BoNT-A, or the combination of the two is superior to the others (105). Lack of evidence was primarily attributed to methodological limitations of the available studies. Strengthening Formalized strength testing in ambulatory children with spastic diparesis or hemiparesis has confirmed greater weakness in all muscles tested using age-matched controls (106). Weakness was more pronounced distally, as expected, and hip flexors and plantar flexors were relatively stronger than their antagonists when compared to the strength ratios of the control group. Strength of the uninvolved side in children with hemiparesis was also weaker than age-matched controls (106). Deficits in voluntary muscle contraction in CP are felt to be due to decreased central nervous system motor unit recruitment, increased antagonist coactivation, and changes in muscle morphology, including muscle fiber atrophy and increased fat and connective tissue (107). This weakness is thought to be a large contributor to functional deficits in children with CP, but historically, strengthening programs were not recommended due to concerns of increasing spasticity. A number of studies have shown, however, that strengthening programs can increase strength without adverse effects such as increased spasticity, resulting in an increased interest in strengthening programs for children with CP (108,109). Strength or resistance training has been the subject of several systemic reviews and has been included in reviews of different therapy approaches to improve gait or other aspects of motor function (109–111). The general consensus across studies is that strength can be predictably increased through a properly designed short-term program, but training likely needs to be continued to retain benefits (109). Strengthening in functional positions may transfer more readily to improvement in motor tasks; however, it is uncertain whether it is the loading or the functional practice that makes the difference (110). Although strengthening has the potential to positively affect children with CP in many areas of the International Classification of Functioning, Disability and Health (ICF) model, most studies have focused on changes in strength alone. Some activity level improvements have been reported with strength training, as measured by the Gross Motor Function Measure (GMFM) (112–114). Other studies have reported an improvement in strength, but no associated improvement in function (115–117). The lack of functional improvement has been attributed to a number of potential causes including: insufficient dose or density to drive neuroplasticity (118), the lack of context-specific training (118), or inadvertently strengthening nondesired muscles, leading to further muscle imbalance (119). Walking is a complex task and there are a number of other impairments besides weakness that can make ambulation difficult including impaired motor planning, impaired balance and postural control, abnormal tone, and decreased ROM (116). Although not typically measured, increased participation, self-esteem, and participant-rated functional mobility have been associated with participation in a strengthening program (116,120,121). Strengthening appears to be a promising intervention for children with CP, but future studies are needed to determine the effect of individual patient factors on a wide variety of potential outcomes, including societal participation. TREADMILL TRAINING (TT) AND PARTIAL BODY WEIGHT SUPPORT TREADMILL TRAINING (PBWSTT) TT is utilized to provide repetitive task-specific walking practice. The goals are to increase lower extremity (LE) strength, increase speed, improve symmetry of gait patterns, or to increase endurance (110). A systematic review and recent studies provide support for improvement in temporal–spatial characteristics of gait, 10-m walk test, and GMFM scores with the use of TT (122–124). Little evidence is available on the effects of TT on participation. To facilitate step training, weight support systems were developed to be used in conjunction with treadmills to reduce postural requirements and thus the amount of physical assistance needed to safely participate in ambulation training (110). A systematic review of PBWSTT also revealed significant improvements in temporal–spatial characteristics of gait and GMFM scores (122). An RCT comparing PBWSTT to overground gait training found no additional benefit with the PBWSTT (125). Robotic-assisted treadmill therapy utilizes two mechanically driven leg orthoses, resulting in a kinematic pattern resembling normal walking. This allows for an intensification of locomotor training by increasing the amount of stepping practice, as well as altering the amount of body weight support being provided while decreasing the therapist’s manual assistance. To date, few studies have reported on the effects of robotic-assisted TT in children with CP. A small noncontrolled study showed improvements in walking and standing performance on the GMFM (126) and a prospective controlled cohort study revealed improvements in activity and participation (127). Larger RCTs will be needed to increase confidence in the positive effects of TT and PBWSTT and to determine practice guidelines. CONSTRAINT-INDUCED MOVEMENT THERAPY (CIMT)/INTENSIVE BIMANUAL TRAINING CIMT and intensive bimanual training are motor-learning-based approaches that focus on upper limb (UL) function in children with hemiplegic CP. CIMT was developed for treating adults with hemiparesis or “learned nonuse” following a stroke (128). The therapy includes intensive motor practice or shaping of the paretic UE combined with restraint of the uninvolved extremity. Classic CIMT is defined as restraint of the unaffected limb in conjunction with at least 3 hours per day of therapy for at least two consecutive weeks, whereas modified CIMT (mCIMT) uses a more child-friendly approach with the use of a mitt, splint, or bandage and varying frequencies/duration of restraint. CIMT focuses on training unilateral dexterity, but children with hemiplegic CP have impairments with coordination of two-handed activities (129). CIMT does not address this impairment directly and thus generalization of the training may not apply (130). This led to the development of intensive bimanual training, which engages the child in activities that require the use of both hands as a means to increase use of the affected extremity. One highly structured form of bimanual training is hand arm bimanual training (HABIT) (131). Sequential use of CIMT followed by intensive bimanual therapy (hybrid therapy) has also been tried in order to obtain the unique benefits of both individual therapies (132). CIMT and intensive bimanual therapy are some of the more thoroughly studied interventions for CP and there is strong evidence for their effectiveness (98). A meta-analysis published in 2013 included 5 classic CIMT studies, 20 mCIMT studies, and 2 hybrid studies (133). The results revealed that individually these studies predominately reported improved UE outcomes, compared with usual care, at substantially lower dosages. The meta-analysis revealed modest to large effect sizes of mCIMT on improving efficiency and quality of movement of the impaired arm compared with usual care. The comparison of unimanual therapy (eg, mCIMT) with an equivalent dose of bimanual therapy yielded similar results. This same study reviewed the success of implementing these therapies in the home setting and found that for mCIMT, between 50% and 80% of the anticipated dose was achieved and 85% for bimanual training suggesting that this form of therapy may be easier to implement in the home setting (133). The preferred frequency, duration, or method of CIMT or bimanual therapy has yet to be determined. An expert consensus panel concluded that future research should be focused on the following three key questions: (a) the effect of age on treatment effect, (b) benefits of repeated treatment sessions, and (c) whether the amount of structured training matters (134). ELECTRICAL STIMULATION Proponents of electrical stimulation suggest that it increases strength and motor function, and it is an attractive alternative for strengthening in children with poor selective motor control (135). Neuromuscular Electrical Stimulation (NMES) NMES utilizes electrical current to produce a visible muscle contraction. The results of two small case series found increased active and passive ROM at the ankle after stimulation of the anterior tibialis (136) and improved sitting balance following stimulation of the abdominal and posterior back muscles (137). Two RCTs failed to identify any statistically significant improvement in strength or function following NMES of the quadriceps (138) or gluteus maximus (139), but both of these studies were underpowered. Functional Electrical Stimulation (FES) If NMES is used to make a muscle contract during a functional activity, it is termed FES. FES is commonly used at the anterior tibialis muscle to increase dorsiflexion during ambulation. A small case series documented improvement in heel strike and ankle dorsiflexion following FES in children (140). Another study identified clinically significant improvements in gait in only 3/8 subjects with CP, as measured by a three-dimensional gait analysis (141). One proposed reason for lack of response was spasticity of the antagonist muscles limiting range and speed of movement. Commercially available devices, commonly referred to as neuroprostheses, are available to deliver asymmetrical biphasic surface electrical stimulation to the common fibular nerve, triggered by a tilt sensor, to improve foot clearance during swing phase. FES may have potential benefits versus traditional orthotic use in foot drop. It does not restrict motion, produces muscle contraction, and thus has the potential to increase strength and motor control (142). It also is likely to be more acceptable from a cosmetic point of view. The results of small studies support its effectiveness and tolerability in managing foot drop in children with CP (142,143). One study reported use-dependent muscle plasticity in CP, but permanent improvements in voluntary ankle control after repetitive stimulation were not demonstrated (144). Some participants had a decrease in active ankle dorsiflexion after using the device and this was felt to be because they had “learned” to let the device take over ankle control (144). A recent review of the five available RCTs to date on FES in CP found that FES was more effective than no FES intervention, but had a similar effect to activity training alone (145). Threshold Electrical Stimulation (TES) TES is a low-level electrical stimulus, often applied during sleep that does not result in a visible muscle contraction. The proposed mechanism of TES is that increasing blood flow during a time of heightened trophic hormone secretion results in increased muscle bulk (146). There have been four RCTs evaluating TES to date, and three of them failed to show any improvement in strength or function (138,147,148). The parents, however, reported a perceived positive effect of treatment in two of the studies (147,148), and a decreased impact on disability as measured by the Lifestyle Assessment Questionnaire was found in the third (138). A systematic review of electrical stimulation in CP concluded that the scarcity of well-controlled trials makes it difficult to support definitively or discard the use of this therapy (135). Another systematic review revealed low quality of evidence to support improved gait and moderate-quality evidence to support improved strength with electrical stimulation (98). Further studies with more rigorous designs, longer follow-up, larger sample sizes of more homogeneous subjects, and clarity in the reporting of stimulation parameters are recommended to clarify the age and type of patient most likely to benefit from this intervention (135). SPEECH THERAPY Involvement of a speech and language pathologist is useful in the assessment of children prior to early intervention or early childhood educational planning. Many children with CP have oromotor deficits, dysphagia, dysphonia, and/or articulation and language deficits. It is essential to recognize these deficits promptly and enroll these children into speech therapy services to address treatment strategies in an effort to correct or improve these concerns. HYPERTONIA MANAGEMENT Hypertonicity affects the majority of children with CP (149,150). It may occur focally in distinct muscle groups, as is often the case in diparesis or hemiparesis, or more globally, affecting the majority of axial and appendicular skeletal muscles. Hypertonicity can result in a number of negative effects. It can interfere with positioning, contribute to the formation of contractures and musculoskeletal deformities, and be a source of discomfort. It can also negatively affect function and make caregiver tasks, such as transfers and dressing, more difficult. Increased tone can sometimes assist with function. For example, increased extensor tone in the lower extremities may assist with standing and transfers. A wide variety of treatment options for hypertonicity are available, including oral medications, nerve blocks, and surgery. Determining whether abnormal tone is present globally or focally and the magnitude of its effect on an individual’s musculoskeletal system, function, and comfort should guide one’s treatment plan. The specific goals of tone reduction should always be determined prior to any intervention. Evaluating for medical causes of increased tone, such as pain or constipation, is very important. The first-line approach should include stretching, splinting, and positioning as appropriate. Other medical or surgical interventions can be used in conjunction with these when further reduction in abnormal tone is desired. CHEMICAL DENERVATION Chemical denervation should be considered for the treatment of significant focal increases in tone. Alcohol Blocks Alcohol nerve and motor point blocks have been used for many years to reduce focal increases in tone. Phenol injections, at 3% to 5% solutions, either at motor points of selected muscles or perineurally, denature proteins and disrupt efferent signals from hyperexcitable anterior horn cells by inducing necrosis of axons (151–153). Alcohol blocks have the potential to cause painful dysesthesias (153). Nerves that are more commonly treated with phenol include the musculocutaneous and obturator nerves, given the reduced sensory function of these nerves and the lower risk for dysesthesias. The low cost of phenol, coupled with reports of duration of action exceeding 12 months (154), renders phenol injections an attractive treatment option in selected patients with focal spasticity (151). They are frequently done under general anesthesia, however, adding additional risks and costs. There is no published evidence to support or refute the use of these blocks in children with CP (98,155). Botulinum Neurotoxin (BoNT) BoNT is a protein composed of a heavy chain, which binds nerve terminals at the neuromuscular junction, and a light chain, which is transported into the nerve terminal blocking the release of acetylcholine presynaptically and thereby weakening the force of muscle contraction produced by the hyperexcitable motor neurons. BoNT exists in seven serotypes, designated A through G. The Food and Drug Administration (FDA) has not approved BoNT for the treatment of spasticity in children. Muscles commonly treated with BoNT include the gastrocsoleus complex, hamstrings, hip adductors, and flexor synergy muscles of the UE. Intramuscular injections can be localized by surface landmarks, electromyographic guidance, and/or ultrasound. Following injection, muscle relaxation is evident within 48 to 72 hours and persists for a period of 3 to 6 months (156). Dosing is based on units derived from the mouse lethality assay and is not equivalent among the various brands. It is dependent upon both body weight and size of the target muscle(s). Universally accepted dosing guidelines do not exist, but consensus statements of dosing and injection techniques are available for guidance (155,157–159). Injections are typically spaced a minimum of 3 months apart due to concerns of antibody formation (151,160). Side effects are rare with BoNT, but may include pain during injection, infection, bleeding, a cool feeling in injected limbs, rash, allergic reaction, flu-like symptoms, excessive weakness, and fatigue (161–163). Although reports of life-threatening side effects are extremely rare following treatment with BoNT, the risk of systemic spread is possible. The FDA sent out an early communication in 2008 about an ongoing safety review of BoNT A, B following reports of reactions that were suggestive of botulism. The most serious cases resulted in hospitalization and death, and occurred most commonly in children with CP treated for limb spasticity. At the time of writing, the FDA has not completed its review, but recommends that all health care professionals who use BoNT should: (a) understand the potency determinations expressed in “units” or “U” are different among the botulinum toxin products; clinical doses expressed in units are not comparable from one botulinum product to the next; (b) be alert to the potential for systemic effects, following administration of botulinum toxins, such as: dysphagia, dysphonia, weakness, dyspnea, or respiratory distress; (c) understand that these effects have been reported as early as 1 day and as late as several weeks after treatment; (d) provide patients and caregivers with the information they need to be able to identify the signs and symptoms of systemic effects after receiving an injection of botulinum toxin; (e) tell patients that they should receive immediate medical attention if they have worsening or unexpected difficulty swallowing or talking, trouble breathing, or muscle weakness (164). Many studies in the literature describe the effects of BoNT-A in children with CP. A number of evidence-based reviews have been published summarizing this vast literature. There is strong evidence that BoNT-A is an effective treatment to reduce spasticity in the upper and lower extremities in children with CP who have localized/segmental spasticity (98,155). There is conflicting evidence regarding functional improvement in children with CP for both the upper and lower extremities (155,157,158). In spite of the lack of clear evidence, published expert opinion and reviews support the use of BoNT-A in the lower extremities in conjunction with physical therapy to improve walking function (98,165). The use of BoNT-A in the UE in combination with occupational therapy to improve function is also supported by expert opinion and reviews in the literature (98,157,166). The use of BoNT-A to improve comfort and ease of care in children with CP who are nonambulant has not been well studied. There has been only one RCT, to date, in this population, which revealed children with CP who received BoNT-A with therapy, versus therapy alone, had better results on the Canadian Occupational Performance Measure, decreased pain, and a corresponding improvement in health status (167). ORAL MEDICATIONS Oral medications are often used as an early treatment strategy for global spasticity. Medications that are most frequently used include baclofen (Lioresal), dantrolene sodium (Dantrium), clonidine, diazepam (Valium), and tizanidine (Zanaflex). All of these medications work through the central nervous system, with the exception of dantrolene sodium and, therefore, have the potential for sedation (Table 14.2). None of these medications have been found to be universally effective in relieving spasticity (168), and evidence related to functional improvement is extremely sparse. The choice of medications is, therefore, often based on the impact of potential side effects on the individual patient. Widely accepted dosing guidelines do not exist for any of these medications for CP and therefore most clinicians utilize a low starting dose followed by a gradual dose escalation to find the best clinical response with tolerable side effects. Benzodiazepines Benzodiazepines have an inhibitory effect on both the spinal cord and supraspinal levels mediated through binding near but not at the gamma-aminobutyric acid (GABA) receptors and increasing the affinity of GABA for GABAA receptors (169). Diazepam is the most frequently used benzodiazepine and oldest antispasticity medication that is still in use (170), but like other oral medications in CP, its effectiveness has not been well evaluated. It is rapidly absorbed, reaching peak drug levels an hour after drug administration. The positive effect of diazepam may be related to general relaxation that permits improvements, especially in those individuals with athetosis and spasticity (171,172). Baclofen Baclofen is a GABA analogue that acts at the spinal cord level to impede the release of excitatory neurotransmitters implicated in causing spasticity (173). Low lipid solubility impedes passage through the blood–brain barrier with more than 90% of the absorbed drug remaining in the systemic circulation (174). As a result, large doses may be necessary to achieve an effect. A study of pharmacokinetics (PK) in children with CP taking oral baclofen found that the PK was largely proportional to dose/kg, making dose adjustments uncomplicated by PK issues (175). Very few studies have been published regarding the use of oral baclofen in CP. Two small double-blind, placebo-controlled, crossover trials produced differing conclusions regarding the effectiveness of baclofen in reducing spasticity, but neither employed validated outcome measures (176,177). Additional studies assessed the effect of oral baclofen for reduction of spasticity and improved function in small numbers of subjects with moderate to severe spasticity. One study showed possible deleterious effects on motor function (178), while the other demonstrated no difference with placebo except in goal attainment (179). Dantrolene Sodium Dantrolene sodium is unique in that it works primarily through actions on the skeletal muscle and not through central nervous system pathways. It inhibits the release of calcium from the sarcoplasmic reticulum, thereby uncoupling electrical excitation from muscle contraction and reducing contraction intensity. It is well absorbed within 3 to 6 hours after ingestion and is metabolized in the liver to 5-hydroxydantrolene, with peak effect in 4 to 8 hours (180). Doses in children range up to 12 mg/kg/day (172). It is often suggested that dantrolene be considered for the treatment of spasticity of cerebral origin because its mode of action is not central nervous system mediated and it is less likely to be sedating (170,172,181), although an expert panel noted that it is rarely used in clinical practice to reduce spasticity in children with CP (155). Their conclusion is that this is most likely due to the lack of evidence in the literature to support its efficacy along with the potential for frequent and serious side effects. Side effects from treatment can include sedation, nausea, vomiting, and diarrhea. Use of dantrolene is also associated with hepatotoxicity (180,182). Liver function studies should be done prior to instituting treatment and periodically while on maintenance therapy (170). There are a few published trials of Dantrium in CP. One report of long-term use of dantrolene in children with spastic diparesis indicated that young children achieved greater levels of function than predicted prior to dantrolene administration and older children were able to move more easily and maintain their highest levels of function (183). Additional oral medications used to treat spasticity in children with CP include alpha 2 agonists, such as clonidine and tizanidine, as well as certain anticonvulsants, including gabapentin (Neurontin). The alpha 2 agonists result in decreased motoneuron excitability by decreasing the release of excitatory amino acids (182). The side effects associated with these agents are frequently the cause of their more limited use and include nausea, vomiting, hypotension, sedation, dry mouth, hallucinations, and hepatotoxicity. In addition, reversible liver enzyme elevations have been noted in 2% to 5% of patients (170). There is little literature to guide the effective use of tizanidine for the management of spasticity in children with CP (155). Gabapentin is structurally similar to GABA, readily crosses the blood–brain barrier, and is not protein-bound. It does not activate GABA, but results in increased brain levels of it (170). Reports of its use in children with spasticity are not available as of yet. TABLE 14.2 MEDICATIONS USED TO TREAT SPASTICITY IN CHILDREN INTRATHECAL BACLOFEN (ITB) ITB was first described by Penn and associates in 1984 and was FDA-approved for the treatment of spasticity of cerebral origin in 1996. Baclofen is delivered directly to the cerebrospinal fluid (CSF) via a catheter connected to an implanted device in the abdomen. The device contains a peristaltic pump, a reservoir for baclofen, and electronic controls that permit regulation of the pump by telemetry. This feature allows baclofen infusion rates to be either continuous throughout the day or at varied dosages in order to accommodate the patient’s specific needs (173). The newer versions of the pump, such as the SynchroMed II (Figure 14.13), have an expected battery life of 5 to 7 years. By infusing baclofen directly into the subarachnoid space around the spinal cord, potentiation of GABA-mediated inhibition of spasticity can be achieved while minimizing side effects related to high levels of baclofen in the brain (151). Administration of intrathecal baclofen produces levels of baclofen in the lumbar CSF that are 30-fold higher than those attained with oral administration (151). The half-life of intrathecal baclofen in the CSF is 5 hours (184).


Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


